
Конспект лекций для студентов всех специальностей дневной и заочной формы обучения Челябинск
| Вид материала | Конспект |
СодержаниеЭнтропия системы. Закон Рауля. Закон Вант-Гоффа. Лекция по теме: «дисперсные системы» |
- Конспект лекций по курсу Начертательная геометрия (для студентов заочной формы обучения, 1032.28kb.
- Краткий конспект лекций Кемерово 2002 удк: 744 (075), 1231.26kb.
- Конспект лекций по курсу "Начертательная геометрия и инженерная графика" Кемерово 2002, 786.75kb.
- Методические указания по подготовке к семинарским занятиям для студентов дневной формы, 1587.03kb.
- Тематический план, рабочая программа и методические рекомендации к семинарским занятиям, 755.58kb.
- Планы семинарских занятий по дисциплине «Экономическая теория» для студентов технических, 527.66kb.
- Конспект лекций для студентов заочной формы обучения по дисциплине " Организация производства", 16.36kb.
- Методические указания для студентов всех специальностей дневной формы обучения Новосибирск, 320.91kb.
- Тематический план, рабочая программа и методические рекомендации к семинарским занятиям, 755.41kb.
- Методические указания к выполнению задания по черчению для студентов всех специальностей, 589.35kb.
Энтальпию образования простых веществ, устойчивых в стандартных условиях, принимают равной 0. Стандартными условиями считают: Т = 298 К, р = 101,3 кПа. Для идеального раствора с = 1 моль/л.
В основе термохимических расчетов лежат:
1) Закон Гесса: тепловой эффект реакции зависит от природы и состояния исходных веществ и конечных продуктов, но не зависит от пути реакции, то есть числа и характера промежуточных стадий.
2) Закон Лавуазье-Лапласа: теплота, выделяющаяся при образовании сложного вещества из простых, равна теплоте, поглощаемой при разложении такого же количества его на составные части.
Расчеты проводят с использованием термохимических уравнений, которые отличаются от химических тем, что в них указаны агрегатные состояния веществ и Теловой эффект реакции. Например, NН3(Г) + НС1(г) = NН4С1(К), ΔН=-176,9 кДж. Здесь г -газообразное, к - кристаллическое, ж - жидкое, тв - твердое агрегатное состояние.
Согласно следствию из закона Гесса ΔН химической реакции равна сумме энтальпий образования продуктов реакции за вычетом суммы энтальпий образования исходных веществ, с учетом стехиометрических коэффициентов. Для уравнения аА + вВ = сС + дД
ΔН°х.р. = ΣΔН°обр.прод. - ΣΔН°обр.исх. = сΔН°с + дΔН°д -(аΔН°А+вΔН°B), кДж.
По этому уравнению можно рассчитать АН°Х.Р., энергию химической связи, энергию кристаллической решетки, теплоту сгорания топлива, калорийность пищи, энергию фазового перехода и полиморфных превращений.
Знание ΔН°Х.Р. позволяет судить о возможности самопроизвольного протекания процесса. Всегда самопроизвольно протекают экзотермические процессы, то есть имеющие ΔН°Х.Р. меньше нуля. Но иногда и эндотермические реакции могут протекать самопроизвольно.
Энтропия системы. Движущей силой самопроизвольного процесса может быть также стремление частиц (молекул, ионов, атомов) к хаотичному движению, а системы - к переходу от более упорядоченного к менее упорядоченному состоянию. Мерой неупорядоченности состояния системы служит термодинамическая функция, называемая энтропией - S, Дж/моль·К. В отличие от других термодинамических функций, можно определить не только изменение, но и абсолютное значение энтропии. Энтропия идеального кристалла при абсолютном нуле равна 0. Этот постулат Планка называют третьим законом термодинамики. Энтропия образования индивидуальных веществ в стандартных условиях обозначается S°298 и ее изменение рассчитывается также, как и для энтальпии химической реакции:
ΔS°Х.Р. = ΣS0прoд. - ΣS°исх. , Дж/К.
Значение ΔS°х.р. может также в какой-то степени позволить судить о термодинамической возможности протекания процесса. Одна из формулировок второго закона термодинамики: в изолированных системах самопроизвольно протекают только такие процессы, которые сопровождаются возрастанием энтропии. Второй закон термодинамики справедлив лишь для систем, состоящих из очень большого числа частиц, то есть имеет статистический характер. Реальные химические системы практически не бывают изолированными, так как они обмениваются энергией с окружающей средой. Таким образом, химические реакции обычно сопровождаются изменением как энтальпии, так и энтропии.
Критерием самопроизвольного протекания процесса является свободная энергия Гиббса ΔG°0бр., кДж/моль. Изобарно-изотермический процесс термодинамически возможен, если происходит уменьшение энергии Гиббса: ΔG °Х.Р. <0. В обратимом процессе условие ΔG °Х.Р. > 0 означает, что процесс пойдет в обратном направлении. ΔG °Х.Р. = 0 - условие термодинамического равновесия. Функция состояния ΔG °Х.Р. объединяет энтальпию и энтропию в уравнение:
ΔG °Х.Р. = ΔН°Х.Р. - Т ΔS°х.р.
Из этого уравнения видно, что самопроизвольно могут протекать и процессы, для которых ΔН°Х.Р. больше нуля, то есть эндотермические. Это возможно, когда ΔS°х.р. > 0, но Т ΔS°х.р. > ΔН°Х.Р., и тогда ΔG°Х.Р.<0. С другой стороны, экзотермические реакции (когда ΔН°Х.Р < 0) самопроизвольно не протекают, если ΔS°х.р. < 0 окажется, что ΔG °Х.Р>0.
Тема: Химическая кинетика и равновесие
План
1. Общие представления о скорости химической реакции
2. Закон действия масс.
3. Зависимость скорости химической реакции от температуры. Правило Вант-Гоффа.
4. Уравнение Аррениуса. Энергия активации химической реакции.
5. Гомогенный и гетерогенный катализ.
6. Химическое равновесие. Константа химического равновесия.
7. Принцип Ле-Шателье.
Общие представления о скорости химической реакции
Химическая кинетика - это учение о скорости химических реакций, основная ее задача - управление химическими процессами с целью обеспечения большей скорости реакции и максимального выхода продукта.
Одним из основных понятий в химической кинетике является скорость реакции. Скоростью химической реакции ύ называют изменение количества реагирующего вещества за единицу времени в единице реакционного пространства.
Реакции могут протекать с различными скоростями. Одни из них проходят почти мгновенно, другие - очень медленно. Скорость реакции определяется прежде всего природой реагирующих веществ. Но реакция, имеющая практически нулевую скорость при комнатной температуре, может протекать быстро при нагревании. Можно ускорить взаимодействие и при помощи катализатора. Скорость химической реакции зависит от условий протекания процесса. Итак, скорость гомогенной химической реакции измеряется изменением концентрации одного из веществ, участвующих в процессе, за единицу времени, концентрация С выражается обычно в моль/л, а время t - в минутах или секундах; поэтому размерность скорости реакции моль/л·мин или моль/л·с.
Изменение концентрации исходных веществ и продуктов реакции во времени представлена графиком:
C

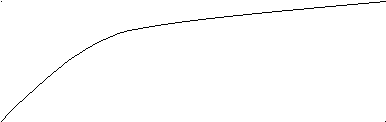 2
21

t
Здесь 1 - изменение концентрации исходного вещества, 2 -изменение концентрации продукта реакции.
При химической реакции концентрации исходных веществ уменьшаются во времени, а продуктов реакции - возрастает. Истинная скорость реакции определяется пределом, к которому стремится отношение ΔС/t при Δt—»0, то есть производной концентрации по времени:
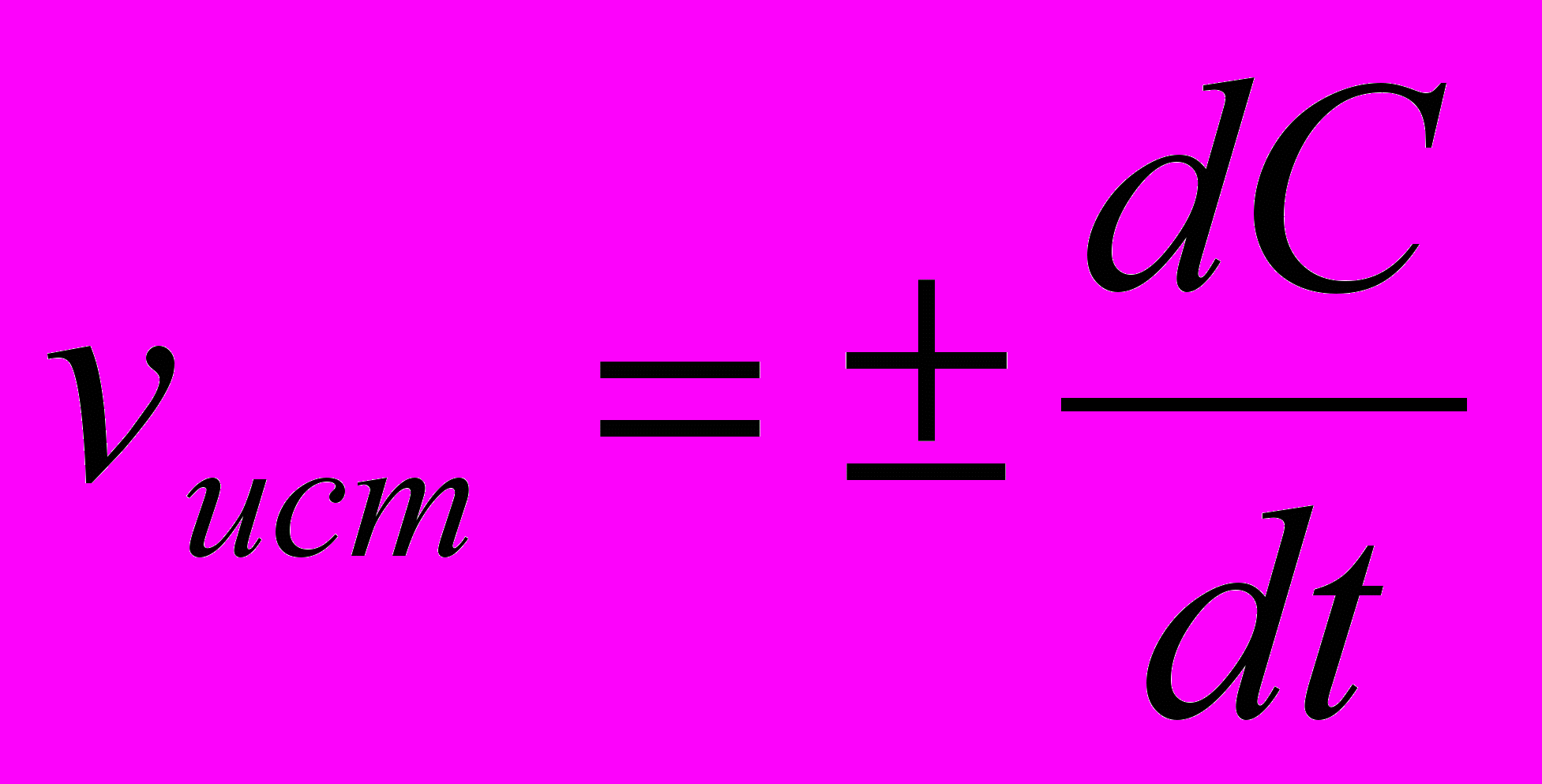 . Если измеряется скорость образования продуктов реакции, то скорость реакции положительна; если измеряется скорость расходования исходных веществ -скорость реакции отрицательна.
. Если измеряется скорость образования продуктов реакции, то скорость реакции положительна; если измеряется скорость расходования исходных веществ -скорость реакции отрицательна.Необходимым условием осуществления химического взаимодействия между двумя молекулами должно их столкновение. Столкновение молекул в некотором реакционном пространстве при заданной температуре происходит тем чаще, чем больше этих молекул. Поэтому скорость химической реакции зависит от концентрации реагирующих веществ. По мере уменьшения концентрации исходных веществ во времени скорость реакции падает.
Закон действия масс
Закон действия масс открыт в 1867 году двумя учеными К. Гульдбергом и П. Ваге. При постоянной температуре скорость химической реакции прямо пропорциональна произведению концентраций реагирующих веществ, взятых в степенях, равных стехиометрическим коэффициентам в уравнении реакции.
Для реакции mА+nВ = С этот закон выражается соотношением ύ=k[A]m[B]n
Здесь [А], [В] - концентрации реагирующих веществ в моль/л, m, n - стехиометрические коэффициенты, k - константа скорости реакции, л/моль·с. При [А]=[В]=1 моль/л ύ = k.
Следовательно, константа скорости выражает собой скорость данной реакции при условии, что концентрации реагирующих веществ равны 1 моль/л. Чем выше величина k, тем быстрее протекает данная реакция. Константа скорости зависит от температуры и природы реагирующих веществ.
Зависимость скорости химической реакции от температуры. Правило Вант-Гоффа.
Скорость химической реакции зависит от температуры, причем при повышении температуры скорость реакции увеличивается. Голландский ученый Вант-Гофф установил, что при повышении температуры на 10° скорость большинства реакций увеличивается в 2-4 раза:
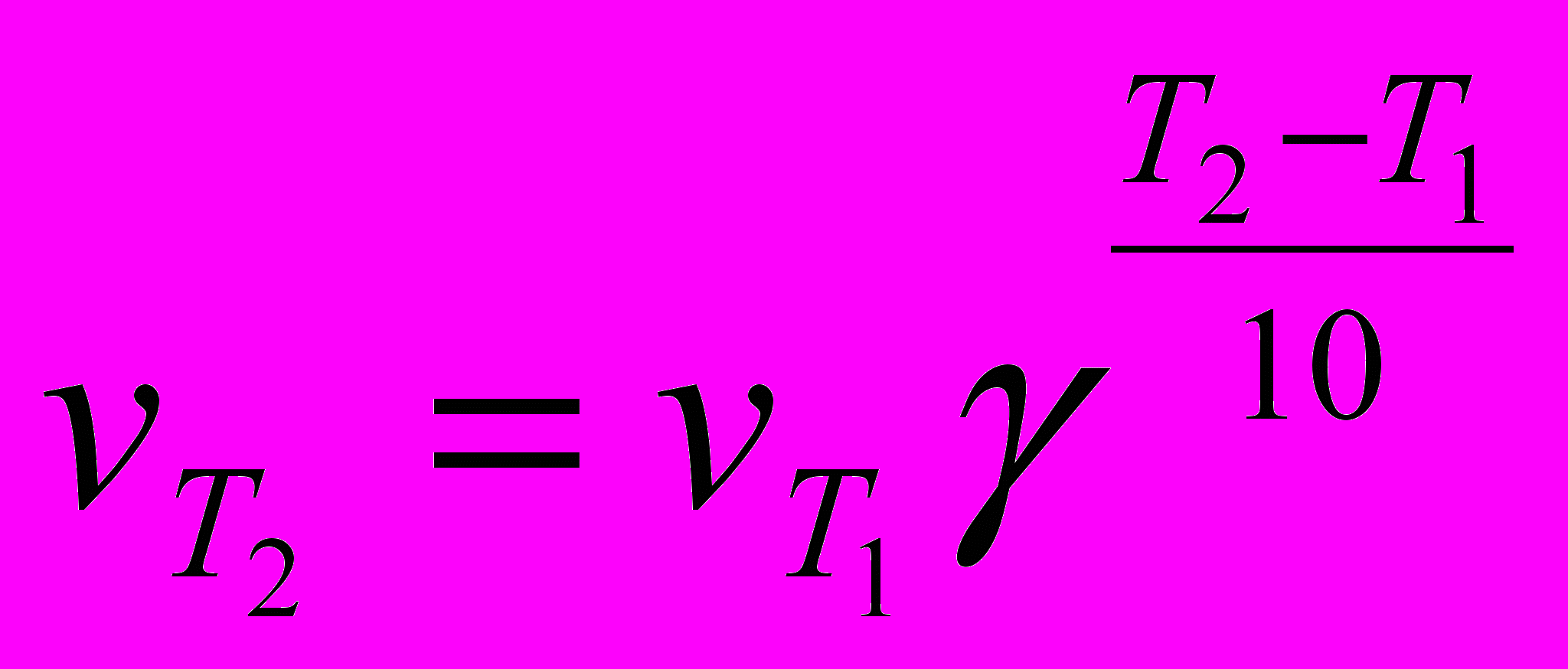
Здесь vT2 и vT1 - скорости реакций при температурах Т2 и Т1, γ -температурный коэффициент скорости реакции. Из уравнения следует, что температурный коэффициент скорости реакции равен соотношению температур, когда Т2-Т1= 10°. Величина γ показывает, во сколько раз увеличится скорость реакции при повышении температуры на 10°. При концентрации реагирующих веществ 1 моль/л скорость реакции численно равна константе скорости, тогда
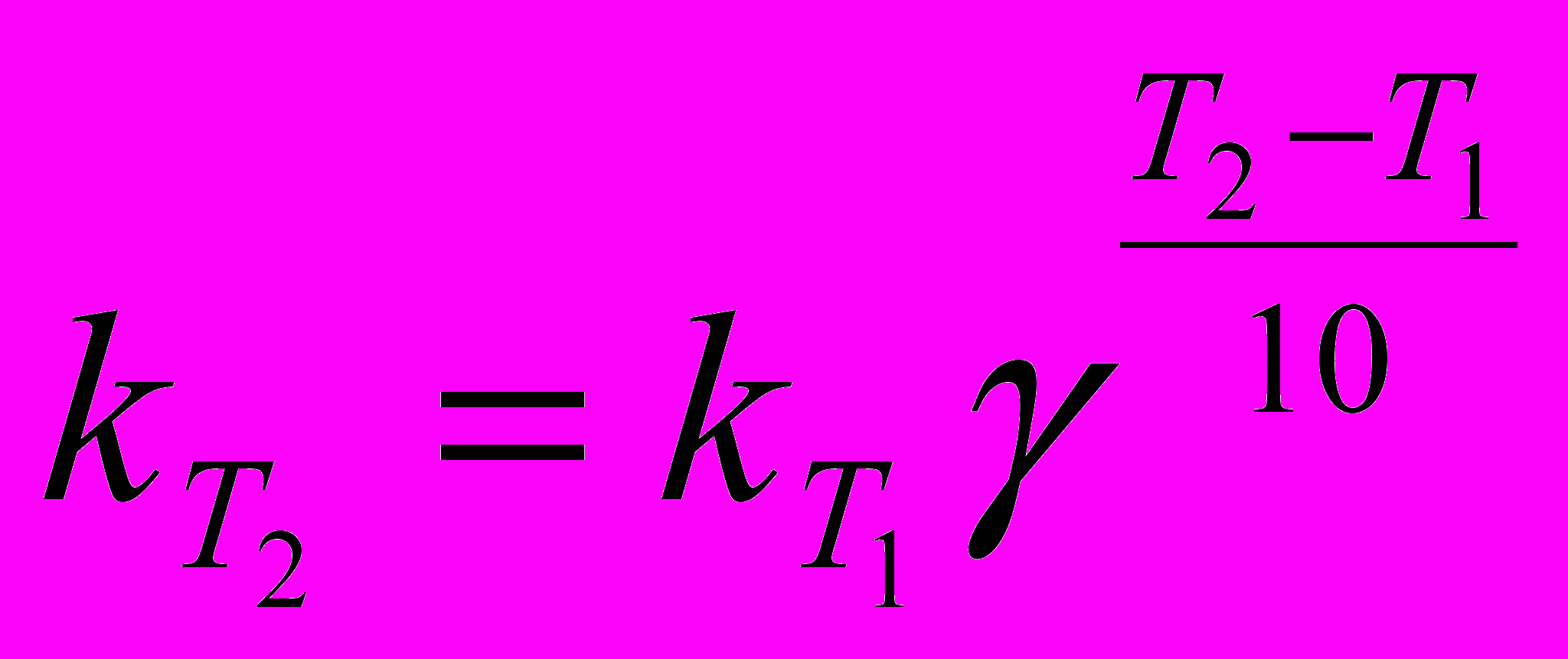 .
.Константа скорости зависит от температуры так же, как и скорость.
Уравнение Аррениуса. Энергия активации химической реакции.
При повышении температуры число активных молекул возрастает и скорость реакции увеличивается. Энергию, которую надо сообщить молекулам реагирующих веществ, чтобы превратить их в активные, называется энергией активации. Ее обозначают Еа и измеряют в Дж/моль.
Величина энергии активации зависит от природы реагирующих веществ и служит характеристикой каждой реакции. Зависимость константы химической реакции от температуры выражается уравнением Аррениуса:
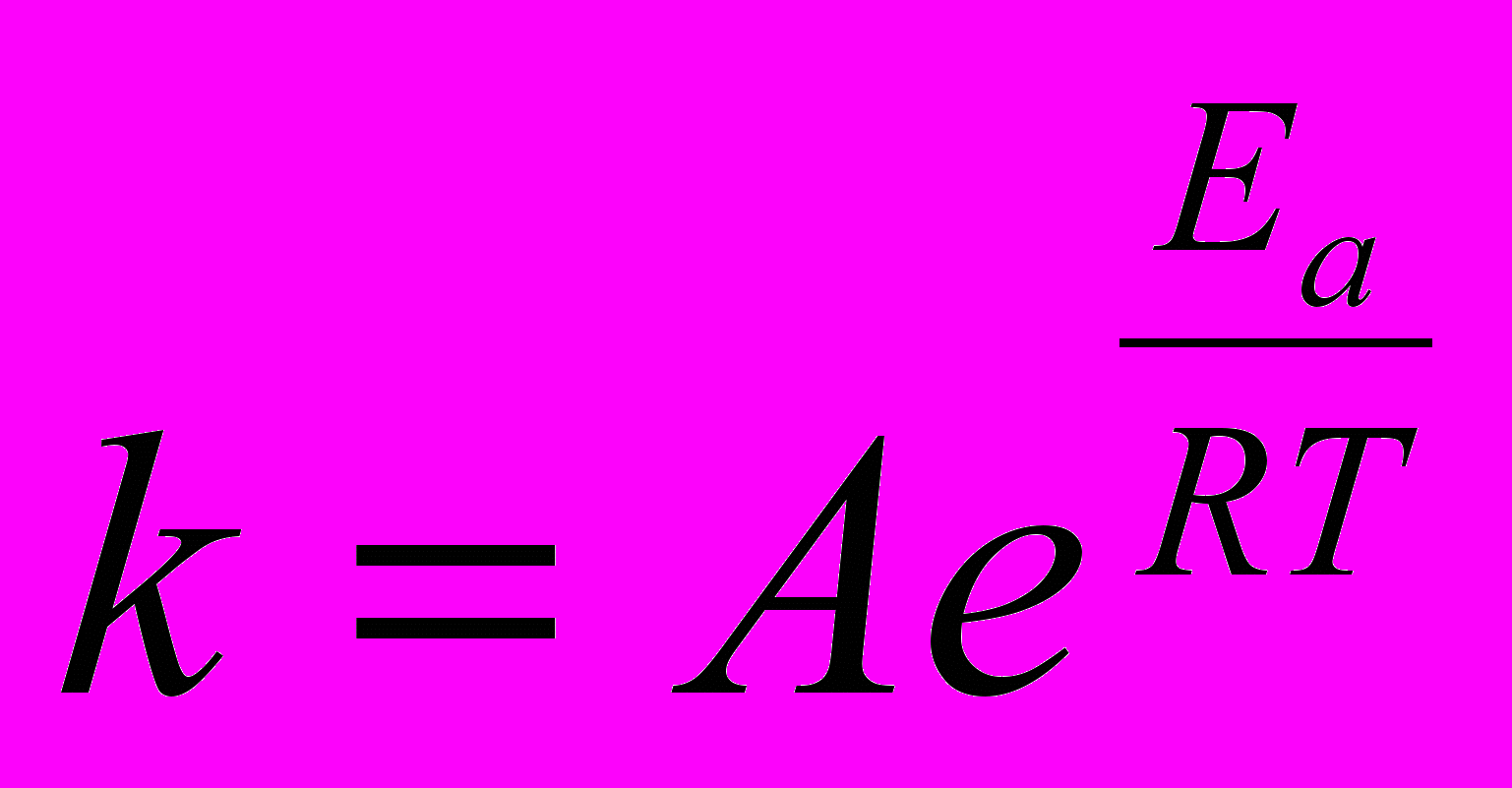
Здесь Еа - энергия активации, Дж/моль, Т - абсолютная температура, К, k - константа химической реакции, R = 8,32 Дж/моль К - универсальная газовая постоянная, А - предэкспоненциальный множитель. После логарифмирования данное уравнение можно привести к виду: ln k = ln A – Ea/RT. Если определить две константы скорости k1 и k2 при температурах Т1 и Т2, то энергию активации вычисляют по формуле
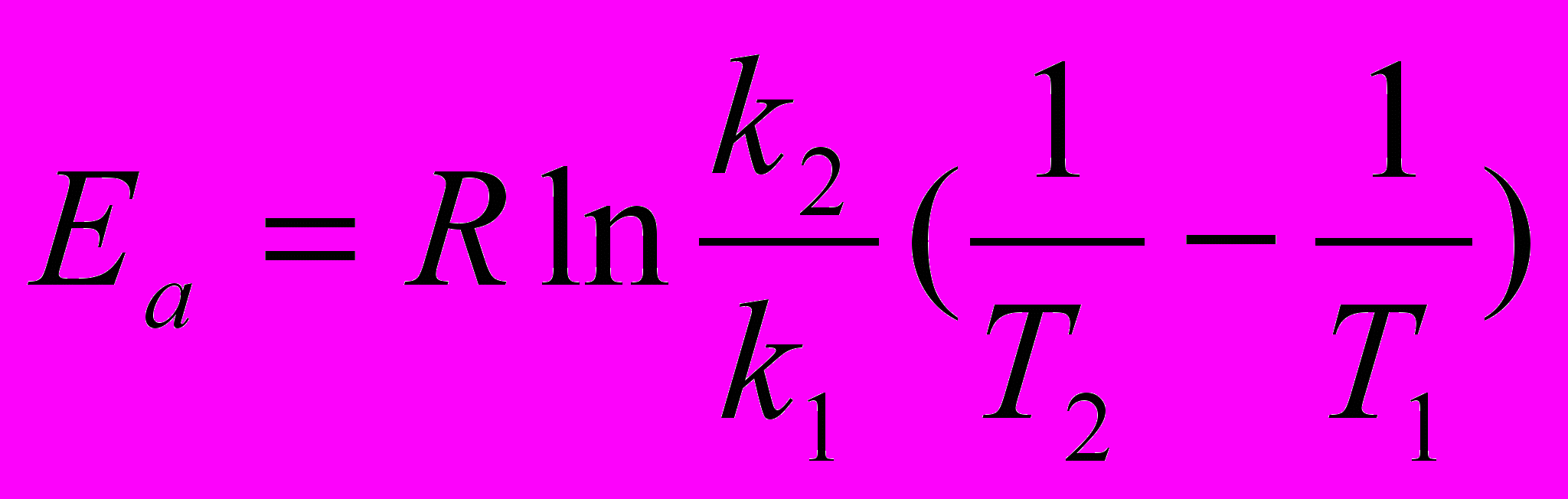
Гомогенный и гетерогенный катализ.
Одним из наиболее распространенным методом ускорения химических реакций является катализ. Этот метод заключается в том, что в реакционную систему добавляют катализаторы - вещества, резко увеличивающие скорость реакции, но не расходующиеся в результате ее протекания. Замедление реакции осуществляется при помощи ингибиторов (отрицательных катализаторов).
Различают катализ гомогенный и гетерогенный. Примером гомогенной реакции является реакция нейтрализация в растворе: НС1+КОН=KС1+Н2О. Примером гетерогенной реакции является термическое разложение карбоната кальция: СаСОз = СаО+СO2.
Скорость гетерогенной реакции зависит от площади контактирующих поверхностей. Если катализатор и взаимодействующие вещества находятся в одной фазе, катализ называется гомогенным. Если катализатор находится в системе в виде самостоятельной фазы - гетерогенным.
Влияние катализатора на скорость реакции при гомогенном катализе объясняется образованием промежуточного соединения катализатора с реагирующим веществом. Так, медленно протекающая реакция А+В=АВ ускоряется катализатором К, вследствие образования реакционноспособного промежуточного соединения А+К=АК, которое взаимодействует с другим исходным веществом В с образованием продукта реакции АВ и выделением катализатора К в первоначальном виде АК+В=АВ+К.
Примером гомогенного катализа может служить окисление диоксида серы в серную кислоту при использовании в качестве катализатора NO2:
2SО2+О2+2Н2О=2Н2SО4.
Эта реакция лежит в основе одного из промышленных методов получения серной кислоты.
Химическое равновесие. Константа химического равновесия.
Реакции, которые при заданных условиях протекают одновременно в двух взаимопротивоположных направлениях, называются обратимыми и обозначаются mА+nВ↔рС+dD. Согласно закону действия масс, скорости прямой и обратной реакций соответственно равны: ύ1 =k1 [A]m[B]n, ύ2 =k2 [C]p[D]d
При достижении равновесия скорость прямого процесса становится равной скорости обратного: ύ= ύ2. Следовательно, k1 [A]m[B]n = k2 [C]p[D]d , обозначив Kp=k1/k2, получим:
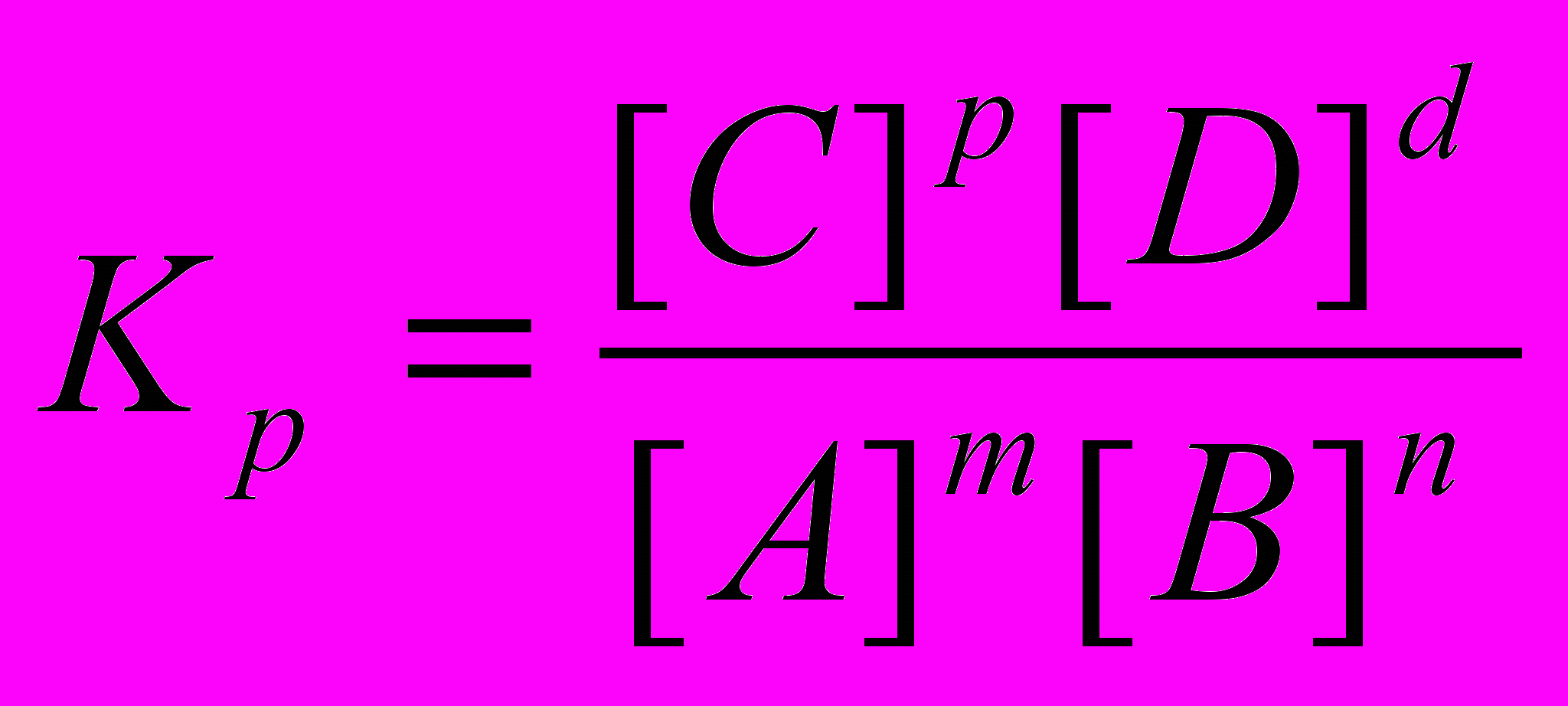 , где Кр - константа равновесия.
, где Кр - константа равновесия.Значение величины Кр дает возможность судить о преимуществе того или иного процесса. Для гетерогенных реакций в выражение для константы равновесия входят концентрации только тех веществ, которые находятся только в газовой или жидкой фазах. Например, для реакции СО2(г)+С(тв)↔2СО(г) КР=[СО]2/[СО]. Константа равновесия зависит только от температуры и природы реагентов.
Изменение энергии Гиббса ΔG обратимого процесса связано с константой химического равновесия следующим уравнением: ΔG = –RТ1nКр. Здесь R - универсальная газовая постоянная, Т- абсолютная температура.
Эта зависимость позволяет, зная ΔG, вычислить константу равновесия и равновесные концентрации реагирующих веществ. Переходя к десятичному логарифму и подставляя значение R, уравнение принимает вид ΔG =-5,71lgКР, кДж/моль.
В условиях химического равновесия концентрации (или парциальные давления в случае газов) исходных веществ и продуктов реакции не изменяются во времени и называются равновесными концентрациями (или парциальными давлениями). Они обозначаются так же, но с индексом р: [СО2]Р, [СО]Р.
Принцип Ле-Шателье.
Химическое равновесие, отвечающее равенству скоростей прямой и обратной реакций, и минимальному значению энергии Гиббса, является наиболее устойчивым состоянием системы при заданных условиях, и остается неизменным до тех пор, пока сохраняются постоянными параметры, при которых равновесие установилось. При изменении условий равновесие нарушается и смещается в ту или иную сторону. Направленные смещения равновесия определяются принципом Ле-Шателье: если на систему, находящуюся в равновесии, оказать внешнее воздействие, то равновесие смещается в том направлении, которое ослабляет эффект внешнего воздействия.
Смещение равновесия может быть вызвано изменением температуры, давления, концентрации одного (или нескольких) реагента.
При повышении температуры увеличивается константа равновесия эндотермического процесса (ΔН>0). Это значит, что при повышении температуры равновесие смещается вправо, в сторону прямой реакции, идущей с поглощением теплоты. Константа равновесия экзотермического процесса (ΔН<0) при повышении температуры уменьшается. Это значит, что при повышении температуры равновесие экзотермической реакции смещается влево.
Смещение равновесия может быть вызвано изменением концентрации одного из компонентов: добавлением вещества в равновесную систему или выводом его из системы. Согласно принципу Ле-Шателье, при увеличении концентрации одного из исходных веществ равновесие смещается в правую сторону, а при увеличении концентрации одного из продуктов реакции - в левую.
Если в обратимой реакции участвует хотя бы одно газообразное вещество, смещение равновесие может быть вызвано изменением давления. Повышение давления при постоянной температуре равносильно сжатию газа, то есть увеличению его концентрации. При увеличении концентрации газообразного компонента скорость реакции в соответствии с законом действия масс возрастает, что приводит к смещению равновесия в направлении уменьшения концентрации газообразного компонента. При понижении давления при постоянной температуре газ расширяется, и его концентрация в системе падает. Это вызывает уменьшение скорости реакции, равновесие смещается в направлении увеличения давления газа. Например, при увеличении давления в системе СаСОз ↔ СаО+СO2 возрастает скорость обратной реакции ύ = k2[СО2], что приводит к смещению равновесия в левую сторону. При понижении давления на ту же систему скорость обратной реакции уменьшается, это приводит к смещению равновесия в правую сторону. В системе 2НС1↔Н2+Cl2 все вещества газообразные, при изменении давления смещения не произойдет, так как обе скорости возрастут одинаково.
Итак, в соответствии с принципом Ле-Шателье при повышении давления равновесие смещается в сторону образования меньшего числа молей газообразных веществ и соответственно в сторону уменьшения давления в системе. Наоборот, при внешнем воздействии, вызывающим понижение давления, равновесие смещается в сторону образования большего числа молей газа, что противодействует внешнему воздействию и вызывает увеличение давления в системе.
Тема: «Растворы. Растворы неэлектролитов»
План
1. Общая характеристика растворов. Растворимость
2. Способы выражения концентрации растворов
3. Общие свойства растворов. Законы Рауля и Вант-Гоффа.
Растворение - сложный физико-химический процесс. В зависимости от природы растворенного вещества и растворителя преобладает либо физическая, либо химическая сторона явления. Физическая - статистическое распределение растворенного вещества (разрушение кристаллической решетки, процесс эндотермический, ΔН1>0). Химическая - гидратация образующихся частиц, процесс экзотермический, ΔН2<0. Знак суммарного процесса растворения определяется соотношением величин ΔН1 и ΔН2.
Очень многие химические реакции, в том числе технические и жизненно важные, протекают в жидких растворах. Растворами называют гомогенные смеси переменного состава, находящиеся в состоянии химического равновесия. Растворы - это как минимум двухкомпонентные системы. Растворитель сохраняет свое фазовое состояние при образовании раствора. Его концентрация по определению выше, чем концентрации других компонентов.
Многие свойства растворов зависят от их концентрации. Концентрацией называется отношение количества или массы вещества, содержащегося в системе, к объему или массе этой системы.
Молярная концентрация вещества В Св – отношение количества вещества (
 , моль) к объему системы (V, л):
, моль) к объему системы (V, л): 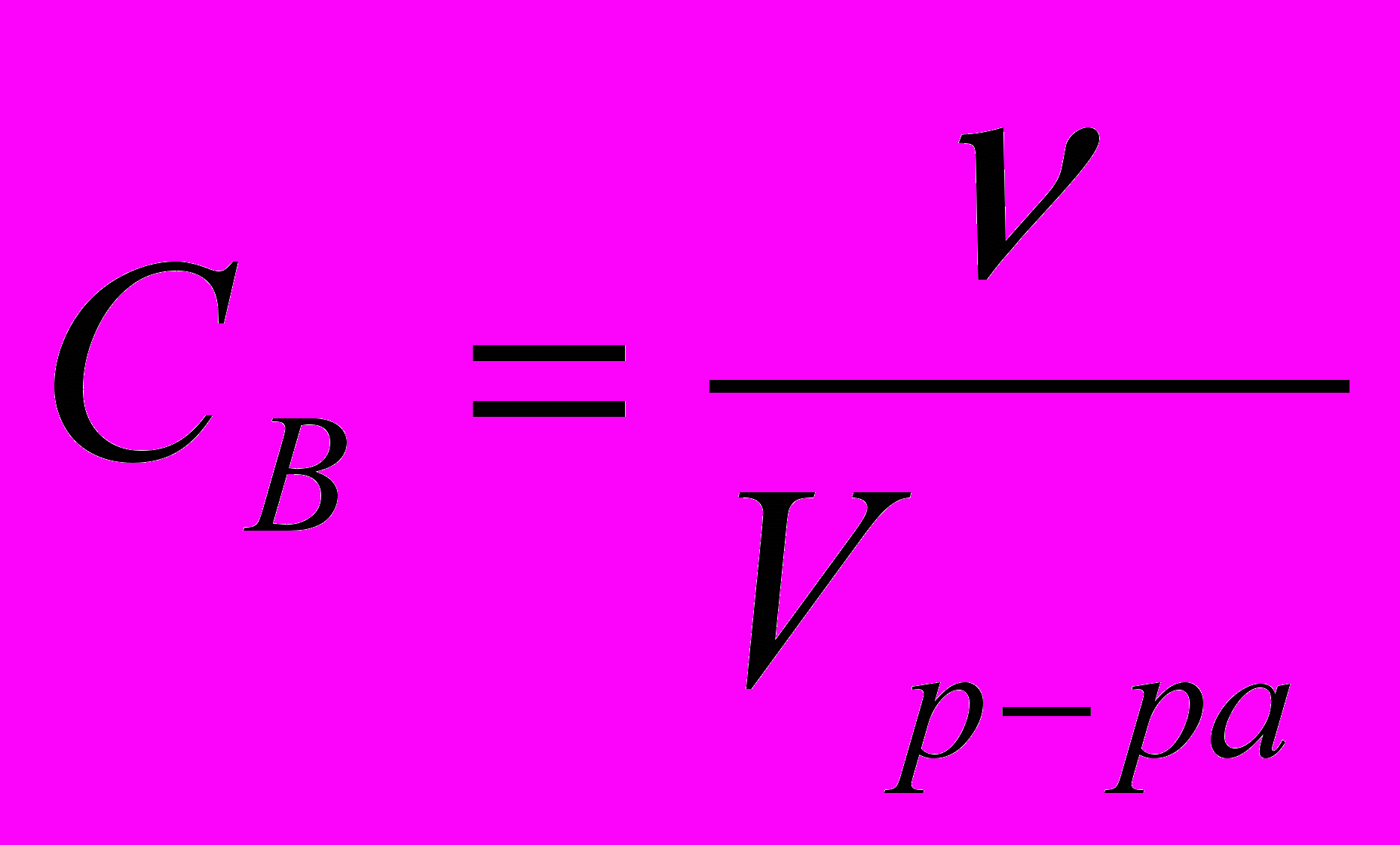 моль/л или моль/м3.
моль/л или моль/м3.Молярная доля вещества В (хв) - отношение количества вещества (моль), содержащегося в системе, к общему количества вещества (моль). Молярная доля может быть выражена в долях единицы и в процентах.
Объемная доля вещества В (φв) - отношение объема компонента, содержащегося в системе, к общему объему системы. Объемная доля может быть выражена в долях единицы и в процентах.
Массовая доля вещества В (ωв) - отношение массы данного компонента к общей массе этой системы. Может быть выражена в долях единицы и в процентах.
Растворимость. При растворении веществ возникает равновесие, при котором скорость растворения фазы равна скорости ее образования.
Растворенное вещество (фаза) ↔ раствор
При равновесии изменение энергии Гиббса равно нулю. Раствор, в котором устанавливается равновесие между растворением и образованием (осаждением, кристаллизацией, выделением вещества), называется насыщенным, а концентрация такого раствора — растворимостью. Растворимость газов уменьшается с увеличением температуры. Растворимость твердых веществ может как увеличиваться, так и уменьшаться, а также мало зависеть от температуры.
Растворимость вещества зависит от свойств растворителя. Вещества с одинаковым характером связей имеют большую тенденцию к взаимной растворимости - подобное растворяется в подобном. Например, полярные и ионные вещества хорошо растворимы в полярных растворителях, неполярные - в неполярных растворителях.
Общие свойства растворов - те свойства, которые не зависят от природы растворенных веществ, а только от их концентрации. Они также называются коллигативными. Такие свойства характерны для идеальных растворов.
Идеальным называют раствор, в котором не происходит химической реакции между компонентами, а силы межмолекулярного взаимодействия между компонентами одинаковы. Энтальпия образования такого раствора равна нулю, каждый компонент является независимым от других. К идеальным растворам по своим свойствам приближаются лишь очень разбавленные растворы.
К общим свойствам растворов относят понижение давления насыщенного пара растворителя над раствором и температуры замерзания, повышение температуры кипения и осмотическое давление. Растворенным веществом в таких растворах является нелетучее вещество, давлением пара которого можно пренебречь.
Закон Рауля. Понижение давления насыщенного пара растворителя А над раствором ΔрА пропорционально молярной доле растворенного нелетучего вещества
хв: р°А – рА = Δр а = р°А хВ,
где р°а, ра - давления насыщенного пара растворителя соответственно над чистым растворителем и раствором. Из уравнения видно, что с увеличением содержания нелетучего растворенного вещества давление пара растворителя над раствором уменьшается, то есть р°а>ра.
Из закона Рауля следует:
1. Температура кипения раствора выше температуры кипения растворителя, потому что давление насыщенного пара растворителя над раствором становится равным атмосферному (условие кипения жидкости) при более высокой температуре, чем в случае чистого растворителя. Повышение температуры кипения пропорционально моляльности раствора:
ΔТкип=КэСm.
Здесь Кэ - эбуллиоскопическая константа растворителя.
2. Температура замерзания (кристаллизации) раствора ниже температуры замерзания чистого растворителя, потому что давление пара растворителя над раствором ниже, чем над растворителем. Понижение температуры замерзания пропорционально моляльности раствора:
ΔТ3ам=КкСm.
Здесь Кк - криоскопическая константа.
Значения эбуллиоскопической и криоскопической констант зависят от природы растворителя; для воды они составляют соответственно 0,52 и 1, 85 кг·К/моль.
Используя следствие из закона Рауля, можно определить молярную массу вещества. Для этого экспериментально определяют изменение температуры кипения или замерзания раствора. Зная массу растворенного вещества mв, его молярную массу находят по уравнению:
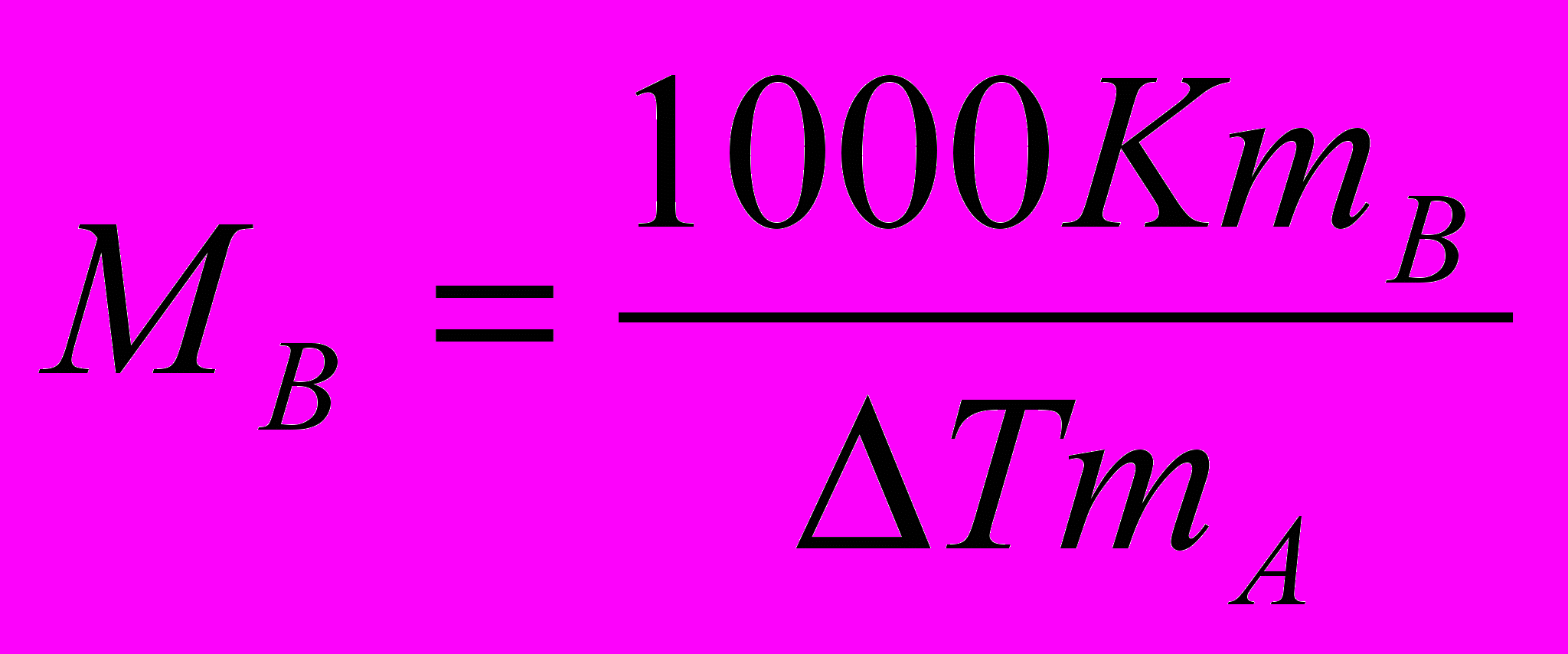 ,
,где К - либо эбулио-, либо криоскопическая константы.
Закон Вант-Гоффа. Самопроизвольный переход растворителя через полупроницаемую мембрану, разделяющую раствор и растворитель, либо два раствора с различной концентрацией растворенного вещества, называется осмосом. Перегородка пропускает только молекулы растворителя, которые переходят из раствора с меньшей концентрацией в более концентрированный раствор. Концентрированный раствор разбавляется и увеличивается высота его столба. Количественно осмос характеризуют осмотическим давлением, равным силе, приходящейся на единицу площади поверхности и заставляющей молекулы растворителя проникать через полупроницаемую перегородку. Осмотическое давление возрастает с увеличением концентрации растворенного вещества и температуры. Вант-Гофф предложил для осмотического давления применить уравнение состояния идеального газа V=nRТ, или =nRТ/V. Отсюда
=СRТ - осмотическое давление.
С -молярная концентрация раствора.
Осмос играет важную роль в биологических процессах, обеспечивая поступление воды в клетки и другие структуры. Растворы с одинаковым осмотическим давлением называются изотоническими; если осмотическое давление выше внутриклеточного, то оно называется гипертоническим, ниже -гипотоническим. Среднее осмотическое давление крови при 36°С равно 78 кПа. Гипертонические растворы, например, соли и сахара (рассол), широко применяют для консервирования овощей (огурцов), так как они вызывают удаление воды из микроорганизмов. Огурцы в крепком рассоле сморщиваются, в гипотоническом растворе - разбухают.
Законы Рауля и Вант-Гоффа соблюдаются лишь в разбавленных растворах неэлектролитов, являющихся моделью идеальных растворов, в реальных растворах вместо концентрации используется активность, о которой подробно будет сказано в следующей лекции.
Тема: «Растворы электролитов»
План
1. Растворы электролитов. Основные положения теории электролитической диссоциации.
2. Степень электролитической диссоциации.
3. Сильные и слабые электролиты. Константа диссоциации.
4. Активность электролитов. Применение законов Рауля и Вант-Гоффа к растворам электролитов.
5. Диссоциация воды. Ионное произведение воды. Водородный показатель.
6. Ионно-молекулярные реакции в растворах. Произведение растворимости.
7. Реакции нейтрализации и гидролиз солей.
Общие свойства растворов электролитов
Вещества, растворы или расплавы которых проводят электрический ток, называются электролитами. Свойства электролитов были рассмотрены и обобщены в трудах Аррениуса, развиты в трудах И.А. Каблукова, В.А. Кистяковского, на основе гидратной теории растворения Д.И. Менделеева. Основные положения теории электролитической диссоциации (ионизации):
1. При растворении солей, кислот и оснований в воде происходит диссоциация (ионизация) этих соединений с образованием электрически заряженных частиц -катионов и анионов.
2. Электрическая проводимость растворов солей, кислот и оснований в воде прямо пропорциональна общей концентрации ионов в растворе.
Уравнение электролитической диссоциации можно записать, опустив промежуточные стадии, указав лишь начальные и конечные продукты реакции:
АВ+(n+m)Н2О ↔ АР+nН2О + Вq–mН2О (1)
Коэффициенты n и m меняются с изменением концентрации, температуры и других параметров раствора. Поэтому молекулы растворителя обычно опускают и записывают: АВ ↔Ap+ + Вq–.
Растворами электролитов являются растворы щелочей, солей и неорганических кислот в воде и других полярных растворителях (жидком аммиаке, ацетонитриле и др.). Растворы электролитов являются ионными проводниками (проводниками второго рода).
В растворах электролитов наблюдаются отклонения от законов Вант-Гоффа и Рауля. По закону Рауля при растворении 0,1 моль неэлектролита (глюкозы) в 1000 г воды температура замерзания снижается на 0,186 К. Поэтому Вант-Гофф ввел в уравнение =СRТ поправочный коэффициент i, который назван изотоническим коэффициентом. Для любых разбавленных растворов Росм=iСRТ. Изотонический коэффициент характеризует отклонение от законов идеальных растворов вследствие электролитической диссоциации.
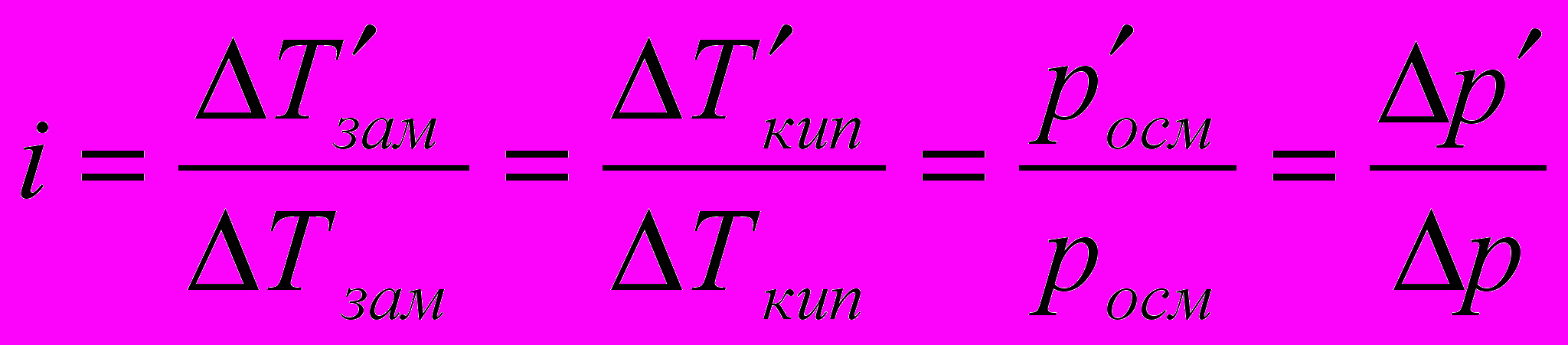
Здесь
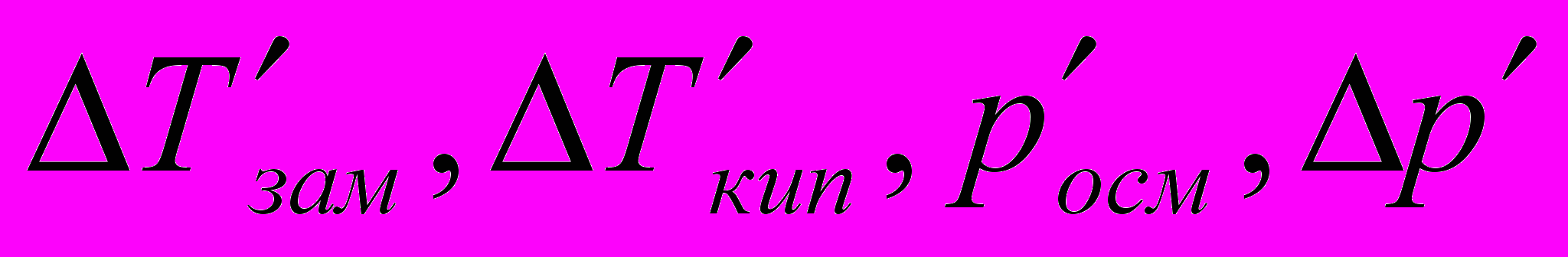 – изменения температур замерзания и кипения, осмотического давления и понижения давления пара для электролита к аналогичному свойству раствора неэлектролита той же концентрации (
– изменения температур замерзания и кипения, осмотического давления и понижения давления пара для электролита к аналогичному свойству раствора неэлектролита той же концентрации (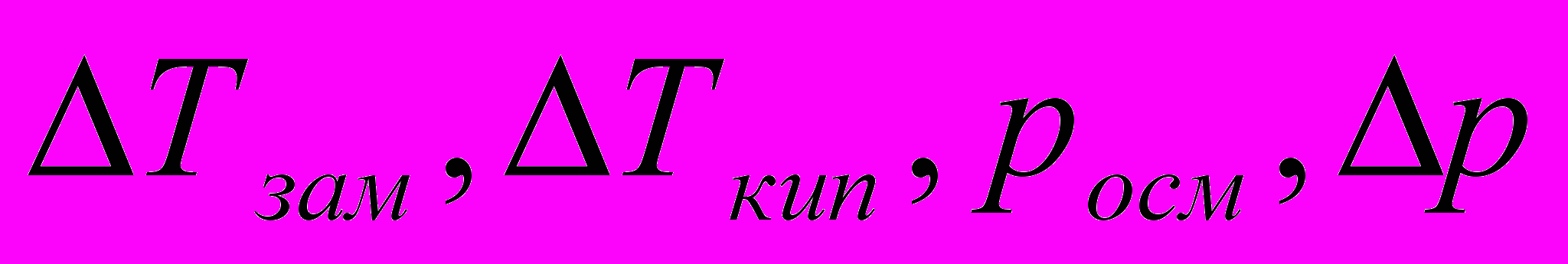 ). Для раствора электролита всегда i>1, для раствора неэлектролита i=1.
). Для раствора электролита всегда i>1, для раствора неэлектролита i=1.Степень диссоциации электролитов.
Отношение числа молекул, диссоциированных на ионы, к общему числу молекул растворенного электролита называется степенью диссоциации (ионизации) ά: ά = n/nо, где n - число частиц, подвергшихся диссоциации, nо - общее число растворенных частиц. По степени диссоциации электролиты делятся на слабые (ά <3%) и сильные (ά >30%). Степень диссоциации зависит от природы (полярности) растворителя. Чем более полярна молекула растворителя, тем при прочих равных условиях выше степень ионизации растворенного вещества. Поскольку электролитическая диссоциация сопровождается тепловым эффектом, то ά зависит от температуры, причем ее влияние можно оценить по принципу Ле-Шателье: если диссоциация представляет собой эндотермический процесс, то с повышением температуры степень диссоциации растет, с понижением температуры уменьшается.
Сильно влияет на степень электролитической диссоциации концентрация раствора. Если рассматривать диссоциацию как равновесный обратимый химический процесс, то в соответствии с принципом смещения равновесия разбавление водой увеличивает количество диссоциированных молекул, то есть степень диссоциации с разбавлением растет.
Константа диссоциации
Процесс электролитической диссоциации удобнее характеризовать константой диссоциации, применив к нему законы химического равновесия. Так, для реакции КА↔К++А– константа диссоциации
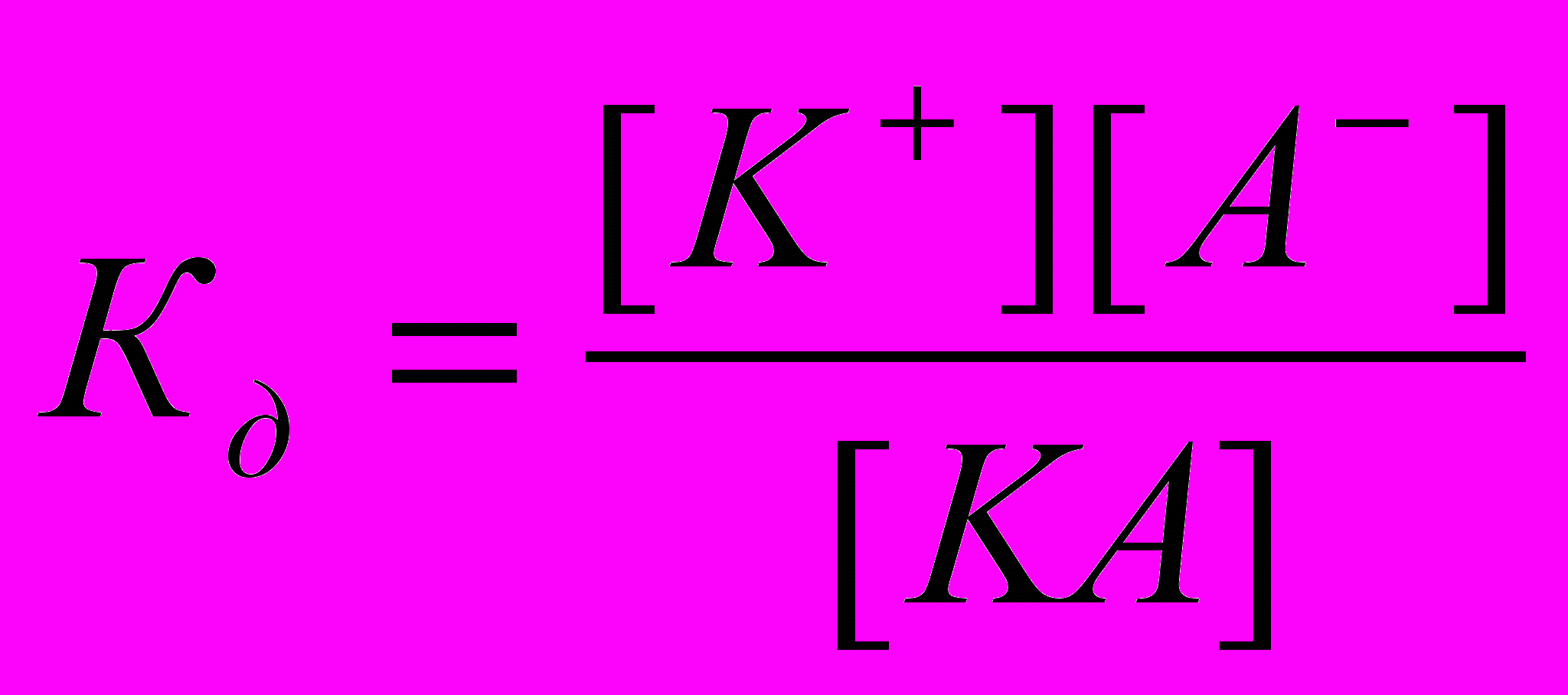 . Здесь и далее в квадратных скобках обозначаются молярные концентрации компонентов. Чем больше
. Здесь и далее в квадратных скобках обозначаются молярные концентрации компонентов. Чем большевеличина
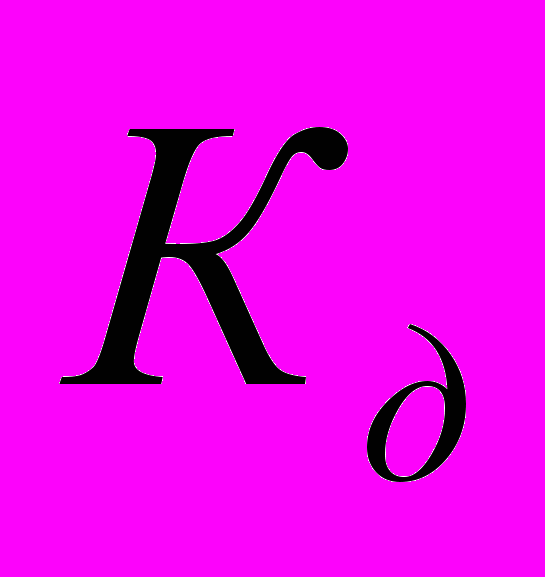 , тем сильнее электролит. Константа диссоциации зависит от природы диссоциирующего вещества и растворителя, а также от температуры, и не зависит от концентрации раствора. Между константой и степенью диссоциации существует количественная зависимость. Пусть в рассмотренном выше процессе диссоциации общее количество растворенного вещества КА равно С, а степень диссоциации ά. Тогда [К+]=[А-]= άС, и, соответственно, концентрация недиссоциированных частиц [КА]= (1- ά)С. Подставив это значение в выражение для константы диссоциации, получим:
, тем сильнее электролит. Константа диссоциации зависит от природы диссоциирующего вещества и растворителя, а также от температуры, и не зависит от концентрации раствора. Между константой и степенью диссоциации существует количественная зависимость. Пусть в рассмотренном выше процессе диссоциации общее количество растворенного вещества КА равно С, а степень диссоциации ά. Тогда [К+]=[А-]= άС, и, соответственно, концентрация недиссоциированных частиц [КА]= (1- ά)С. Подставив это значение в выражение для константы диссоциации, получим: 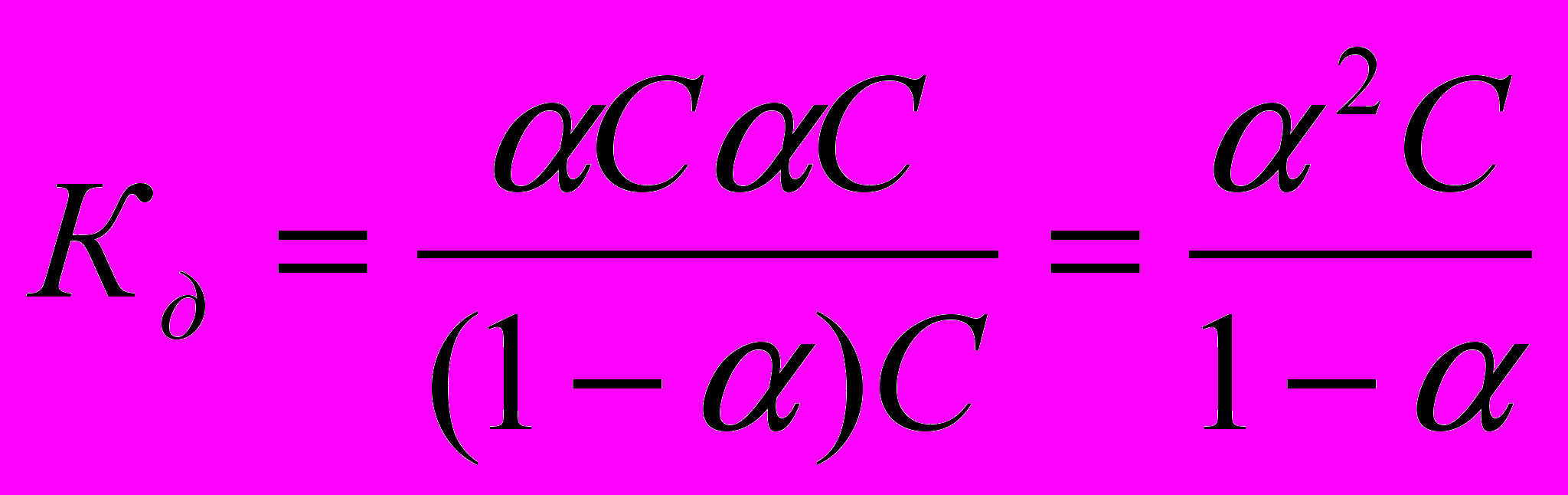 .
.Это соотношение называется законом разбавления Оствальда. Для слабых электролитов, когда ά много меньше 1,
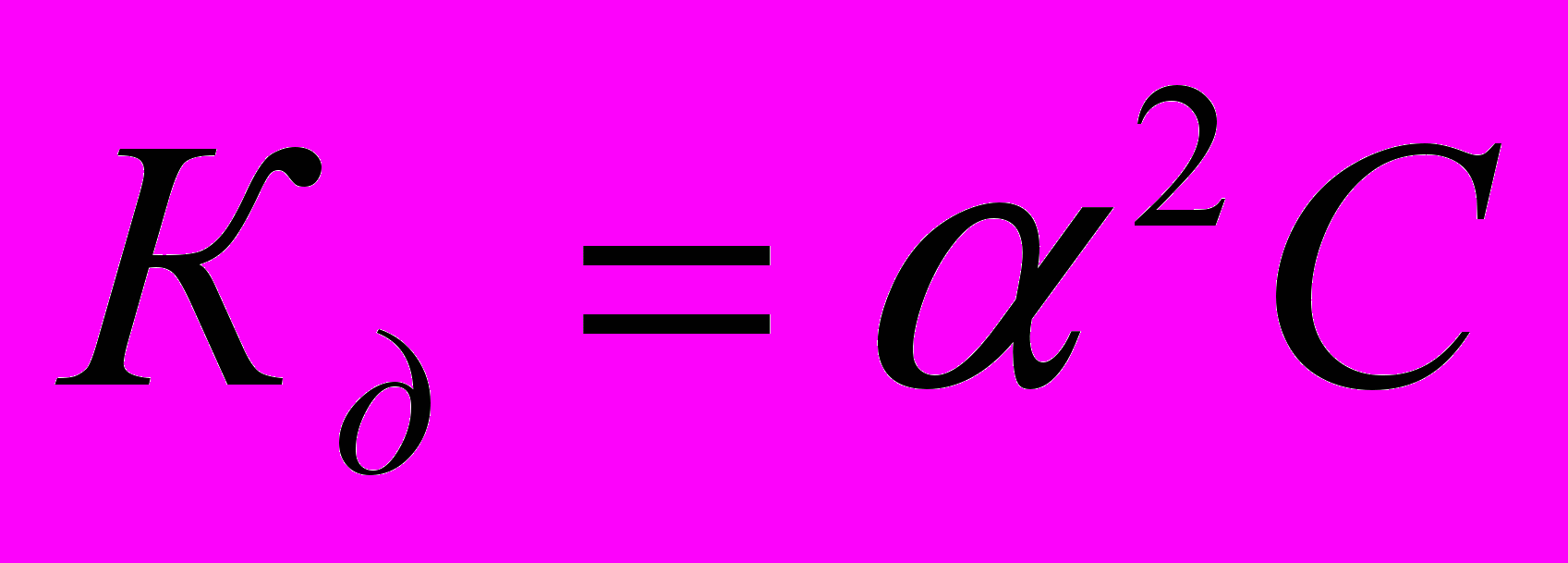 . Отсюда
. Отсюда 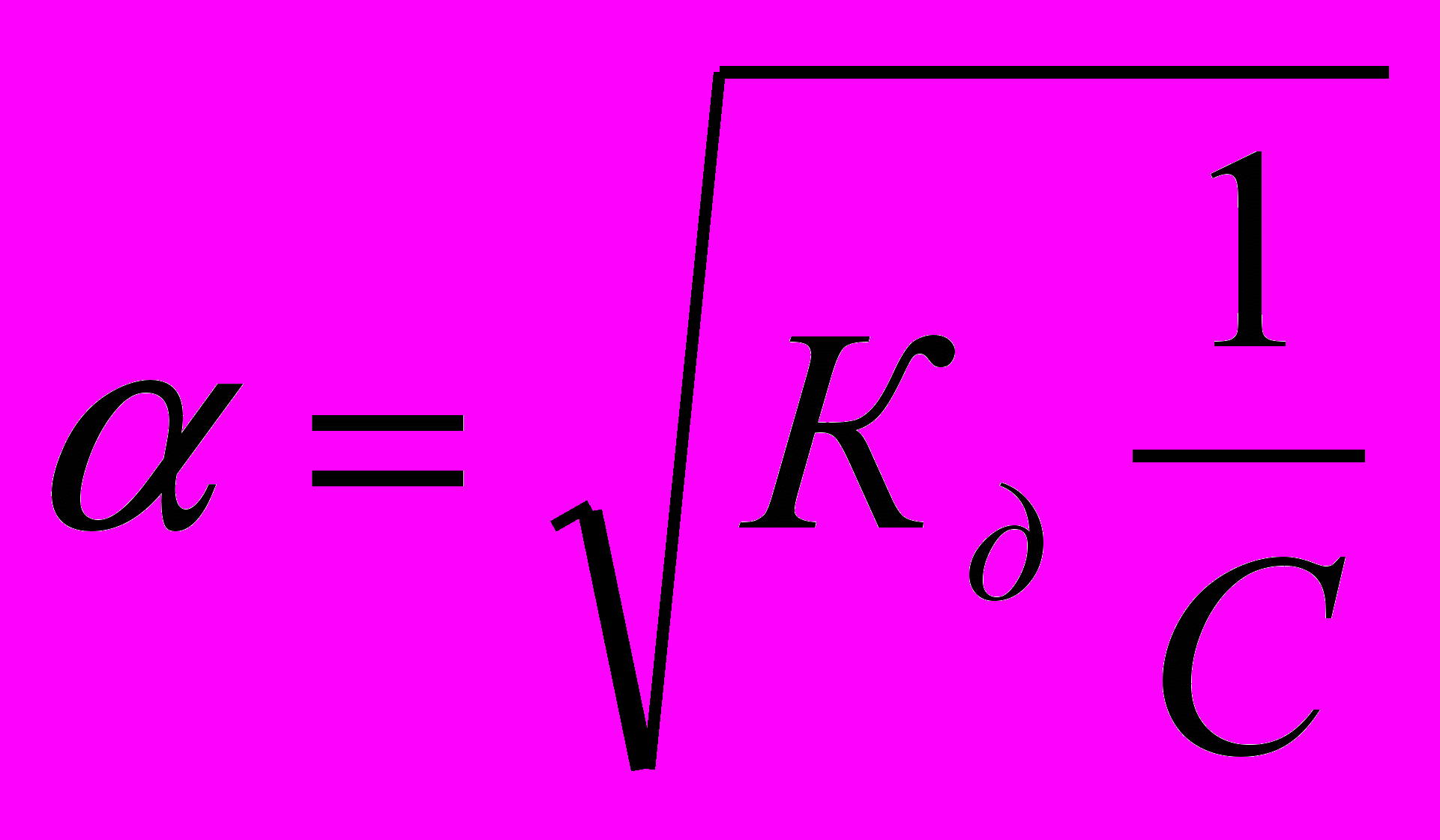 , где V=1/С - разбавление.
, где V=1/С - разбавление.Из уравнения Оствальда следует, что ά уменьшается с увеличением концентрации слабого электролита. Например, при разбавлении раствора в 1 00 раз степень диссоциации возрастает в 10 раз.
В растворах слабых электролитов взаимодействие ионов друг с другом относительно невелико, вследствие их незначительной концентрации. Сильные электролиты в растворах диссоциированы практически полностью, поэтому концентрация их ионов велика и свойства таких растворов зависят от степени взаимодействия образующихся ионов друг с другом и с полярными молекулами растворителя. За счет этого взаимодействия создается впечатление, что диссоциация прошла и в растворе имеется некоторое количество недиссоциированных молекул. Поэтому ά, определяемая в растворах сильных электролитов экспериментально, является кажущейся и ее значение менее 100%. Поэтому для растворов сильных электролитов не применимы законы идеальных растворов. Чтобы эти законы использовать для описания реальных растворов, Льюис ввел представление об эффективной концентрации - активности а: а=γС, где С - концентрация, γ - коэффициент активности (величина безразмерная). Активность и концентрация измеряются в одних и тех же единицах. Коэффициент активности характеризует степень отклонения свойств данного раствора от свойств идеального. Для бесконечно разбавленных растворов электролитов, где практически отсутствует взаимодействие ионов, а=С и γ=1. Если вместо С в уравнения Вант-Гоффа, Рауля, Оствальда подставлять экспериментальные значения а, то уравнения остаются справедливыми и для сильных электролитов, и вообще для любых реальных растворов.
Вода служит не только наиболее распространенным растворителем, но и сама является идеальным амфолитом. Процесс диссоциации воды называется самоионизацией или автопротолизом, и может быть записан: Н2О+Н2О↔Н3О++ОН–. Часто этот процесс записывают в более простом виде: Н2О↔Н++ОН–. Константа диссоциации этого процесса
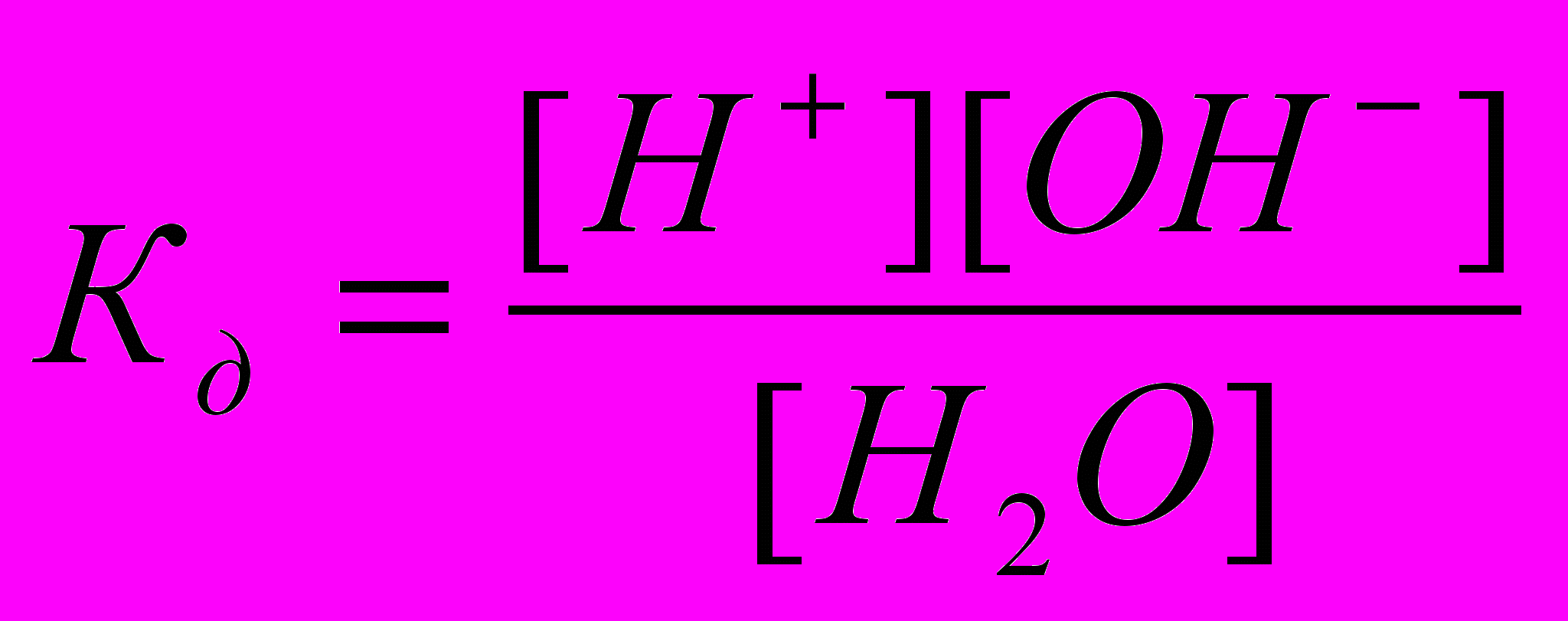 , более строго через активности ионов:
, более строго через активности ионов: 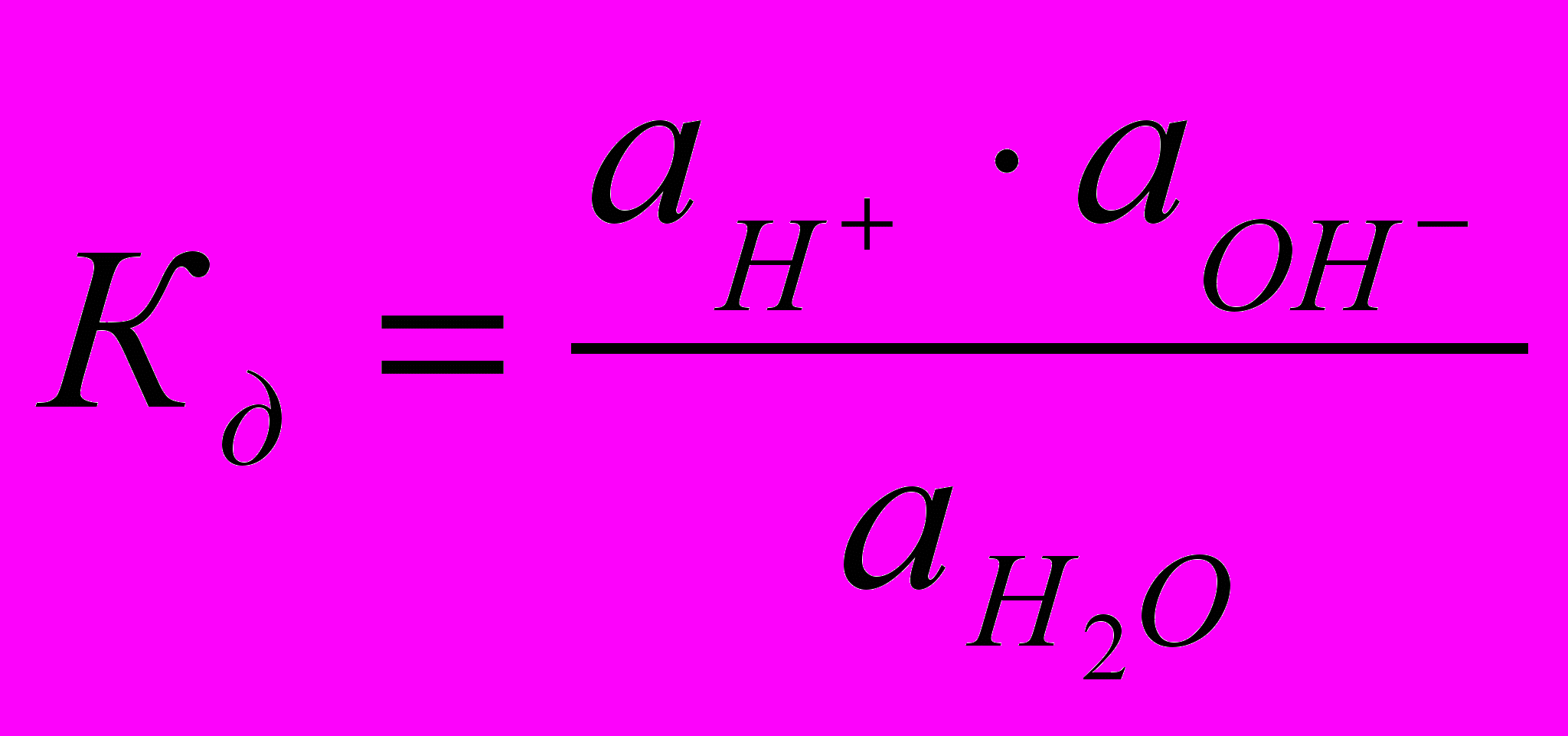 .
.Так как вода - слабый электролит, можно использовать данные выражения с учетом того, что концентрация воды постоянна: [Н2О]=55,56 моль/л. Тогда [Н+][ОН-]=55,56
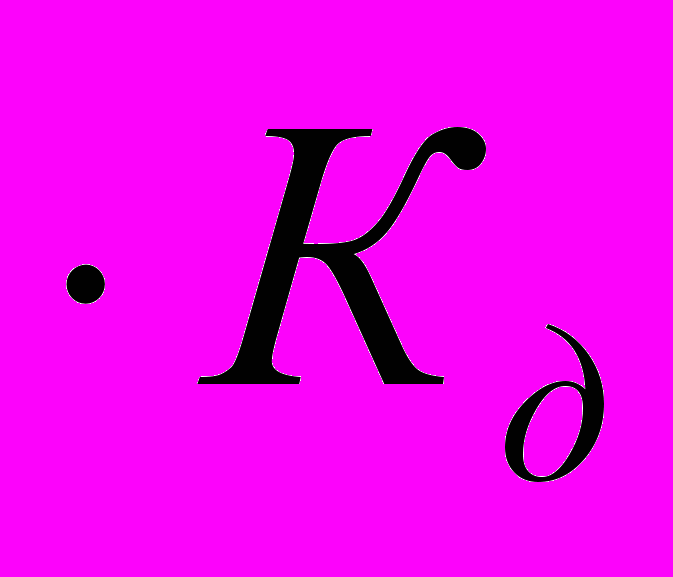 =КW=10-14 – ионное произведение воды. Так как при диссоциации воды [Н+]=[ОН-]=
=КW=10-14 – ионное произведение воды. Так как при диссоциации воды [Н+]=[ОН-]= 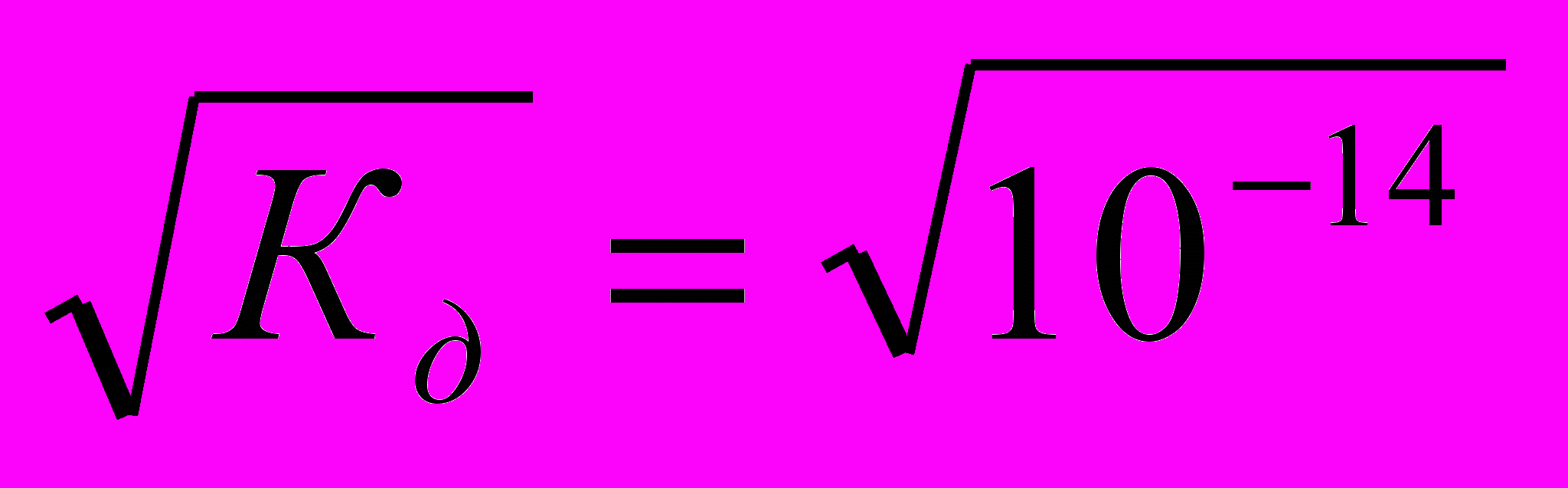 =10-7 моль/л.
=10-7 моль/л.Ионы водорода являются носителями кислотных свойств, гидроксил-анионы - щелочных. Поэтому при равенстве этих ионов раствор будет нейтральным. Для характеристики кислотности (щелочности) среды введен водородный показатель рН=-lg[Н+]. При рН=7 среда нейтральная, для кислых растворов рН меньше 7, для щелочных рН больше 7.
Аналогичным образом реакция среды может быть охарактеризована гидроксильным показателем рОН=-lg[ОН–]. Для воды рН=рОН=7, рН+рОН=14.
Рассмотрим обменную реакцию КС1+АgNОз=АgС1+КNО3. Равновесие в такой реакции смещено вправо, так как хлорид серебра - малорастворимое соединение. Для насыщенного раствора АgС1, находящегося в равновесии с твердой фазой (осадком), будет характерен следующий процесс: АgCl↔Аg++С1–. Константа равновесия этого гетерогенного процесса Кр=Пр= [Аg+][С1–]. Она не зависит от концентрации твердой фазы (АgС1), является постоянной величиной при данной температуре. Эту величину называют произведением растворимости Пр. Более строго в Пр используют не концентрации, а активности ионов электролита Пр=
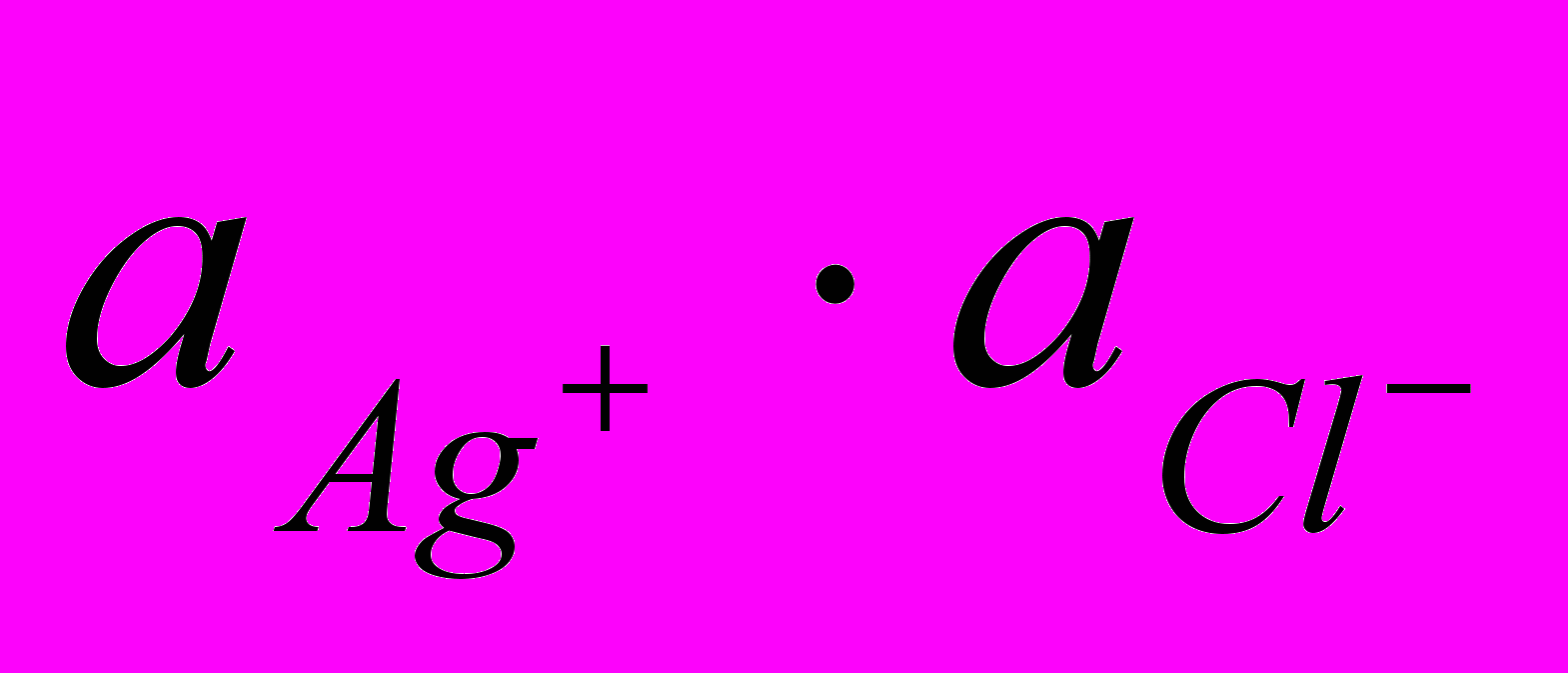 . Применение понятия о Пр аналогично использованию КW. КW определяет ионное равновесие для малодиссоциированного слабого электролита, а произведение растворимости описывает описывает аналогичный процесс для сильного, но малорастворимого соединения.
. Применение понятия о Пр аналогично использованию КW. КW определяет ионное равновесие для малодиссоциированного слабого электролита, а произведение растворимости описывает описывает аналогичный процесс для сильного, но малорастворимого соединения.Реакции нейтрализации и гидролиз солей
К обменным реакциям в растворах относят взаимодействие между кислотами и основаниями, в результате которых образуется соль и вода. Такие реакции называются реакциями нейтрализации и доходят до конца только тогда, когда единственным малодиссоциированным веществом в системе является вода, например: НС1 + КОН = КС1 + Н2О. Сокращенное ионное уравнение этого процесса Н++ОН–=Н2О. Оно показывает, что равновесие полностью смещено в сторону образования воды. Взаимная нейтрализация кислот и оснований, отличающихся друг от друга по силе, до конца не протекает. Так, при взаимодействии слабой уксусной кислоты с гидроксидом калия в системе из четырех веществ два слабодиссоциированы: СНзСООН + КОН ↔ Н2О + СНзСООК, или в сокращенном ионном виде СНзСООН + ОН– ↔ Н2О + СНзСОО–. Эта реакция обратима. Обратная реакция - взаимодействие соли и воды - называется реакцией гидролиза. Гидролизу подвергаются соли, образованные:
1) Слабым основанием и сильной кислотой;
2) сильным основанием и слабой кислотой;
3) слабыми основанием и кислотой.
Соли, образованные сильным основанием и сильной кислотой, гидролизу не подвергаются.
Приведем примеры реакций гидролиза.
Гидролиз соли, образованной сильным основанием и слабой кислотой - ацетата натрия. Соль в растворе полностью диссоциирует: СНзСООNa ↔ СНзСОО– + H+.
Гидролиз в ионной форме: СН3СОО-+Н2О↔СН3СООН+ОН-.
В результате гидролиза реакция среды стала основной (избыток ОН-), рН>7. Показателем глубины протекания гидролиза является степень гидролиза β:
β=Сгидр/С,
где Сгидр – концентрация гидролизованных молекул, С – исходная концентрация растворенных молекул соли. Гидролиз – процесс эндотермический, поэтому, в соответствии с принципом Ле-Шателье, с повышением температуры степень гидролиза увеличивается.
Принимая, что в разбавленных растворах активность ионов мало отличается от их концентрации (С=а), можно записать константу равновесия
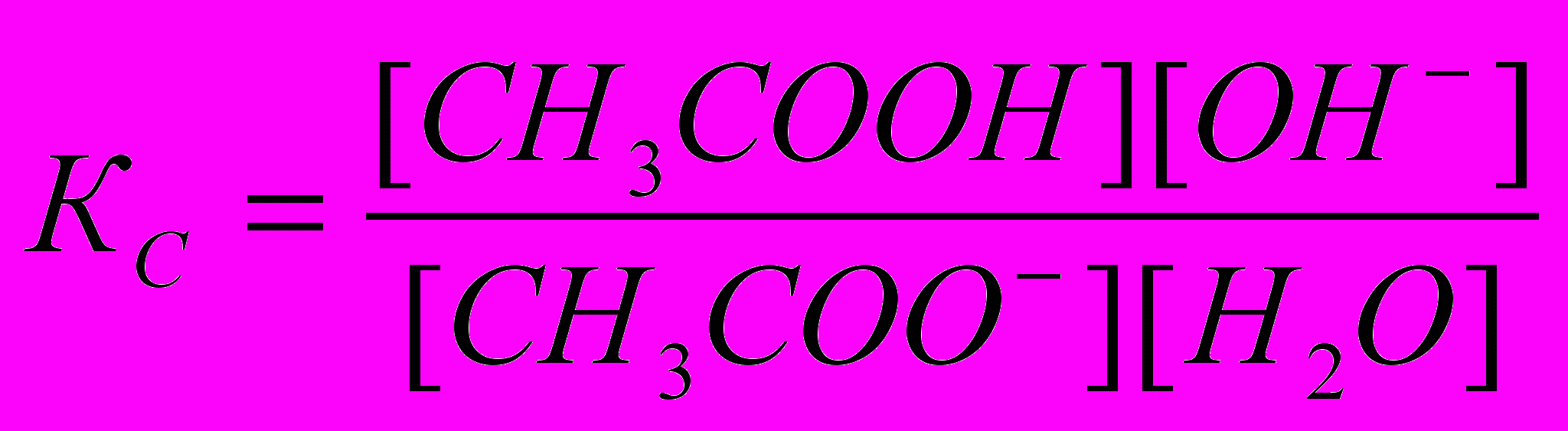 . Так как концентрация воды при гидролизе изменяется мало, принимаем ее постоянной, и, Кс[Н2О]=КГ, получаем константу гидролиза:
. Так как концентрация воды при гидролизе изменяется мало, принимаем ее постоянной, и, Кс[Н2О]=КГ, получаем константу гидролиза: 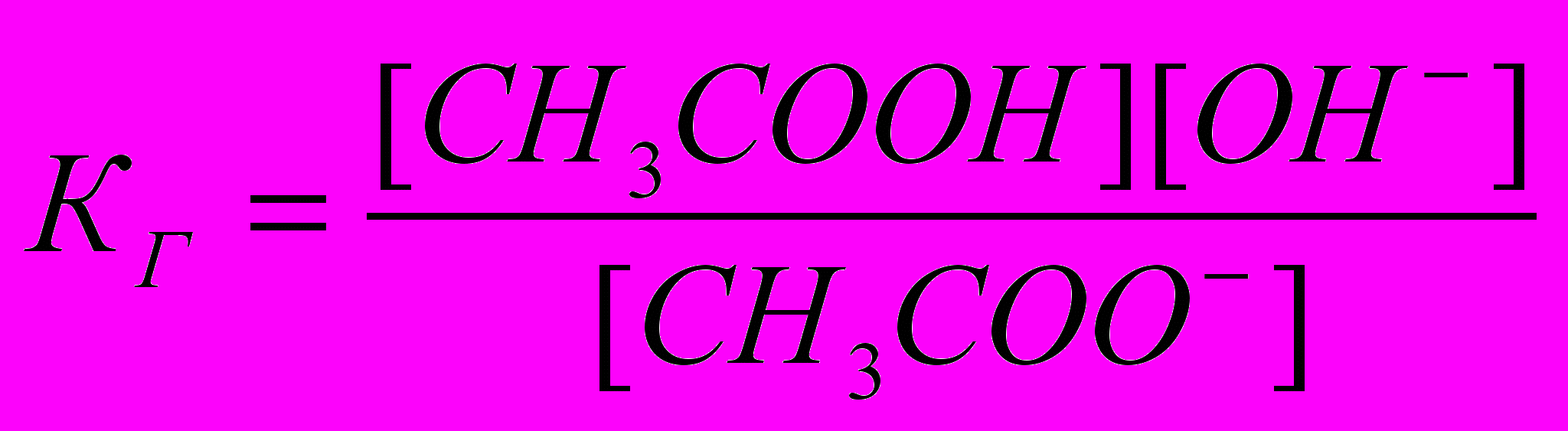 . Подставляя вместо [ОН-]=Kw/[H+], получаем:
. Подставляя вместо [ОН-]=Kw/[H+], получаем: 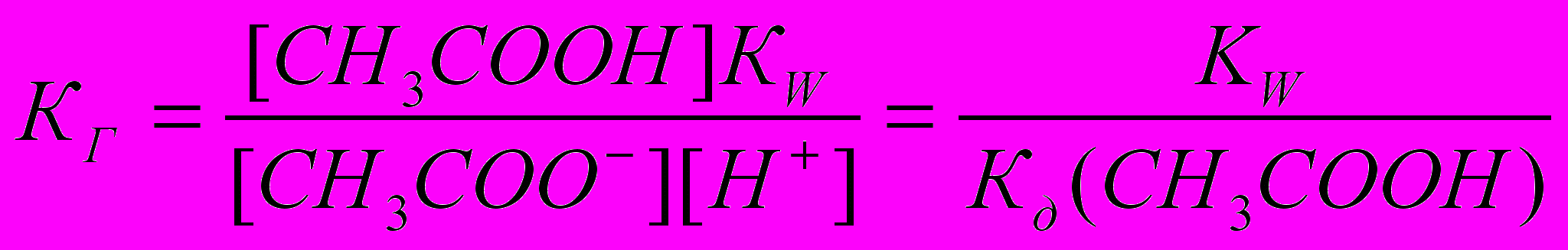 .
.Отсюда следует, что гидролиз идет тем глубже, чем ниже константа диссоциации слабого электролита, образующегося при гидролизе.
Гидролиз солей, образованных сильной кислотой и слабым основанием, например, хлорида аммония:
NH4Cl↔NH4+ + Cl- (диссоциация соли)
NH4+ + H2O ↔ NH4OH + H+, среда кислая, рН<7.
Степень гидролиза и константа гидролиза для данной реакции описываются аналогично приведенным для реакции гидролиза соли, образованной слабой кислотой и сильным основанием.
Гидролиз соли, образованной слабой кислотой и слабым основанием, протекает как по катиону, так и по аниону. Константа гидролиза зависит от констант диссоциации слабых кислоты НА и основания ВОН:
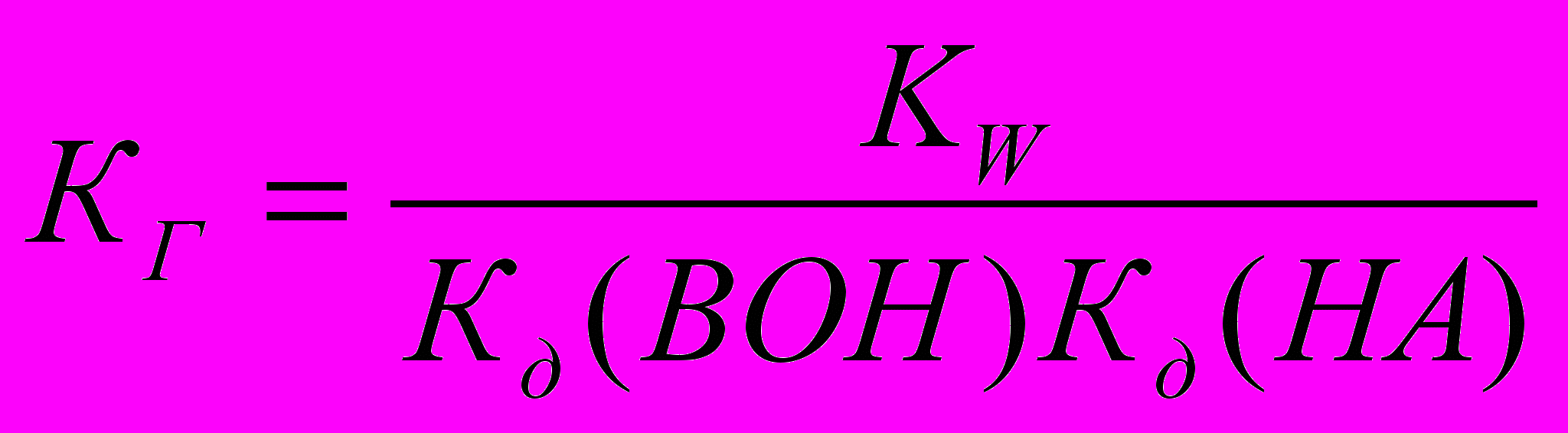 . В зависимости от силы кислоты и основания реакция среды может быть как щелочной, так и кислой.
. В зависимости от силы кислоты и основания реакция среды может быть как щелочной, так и кислой.Итак, гидролиз изменяет реакцию среды (подкисляет или подщелачивает). Степень гидролиза возрастает с разбавлением раствора и при увеличении температуры.
ЛЕКЦИЯ ПО ТЕМЕ: «ДИСПЕРСНЫЕ СИСТЕМЫ»
- Понятия о дисперсных системах, их классификация.
- Коллоидные растворы.
- Строение и заряд коллоидной частицы. Мицелла
- Оптические свойства коллоидных растворов
- Электрические свойства коллоидных растворов
- Коагуляция коллоидных растворов
- Коллоидные растворы в природе и технике
Все дисперсные системы состоят из сплошной фазы, называемой дисперсионной средой, и прерывистой фазы (частиц), называемой дисперсной фазой.
От линейных размеров частиц дисперсной фазы зависит гомогенность или гетерогенность дисперсной системы. Гомогенные дисперсные системы называются истинными растворами, или просто растворами. Истинные растворы содержат молекулы или атомы, размеры которых не превышают 5 нм (5˙10-9 м).
Гетерогенные дисперсные системы подразделяют на: а) грубодисперсные системы, у которых частицы имеют размер 1000 нм (10-6 м) и более; б) коллоидные системы, размер частиц которых лежит в пределах от 1 до 500 нм (10-9 до 5˙10-7 м).
К грубодисперсным системам относятся суспензии, эмульсии, пены.
Суспензии представляют собой системы, состоящие из раздробленного твердого вещества и жидкости, в которой распределена дисперсная фаза. Например, крахмал в холодной воде, шоколад (какао в масле), мутная вода. Примеры концентрированных суспензий – пасты, взвесь глины в воде.
Эмульсии – образуются двумя несмешивающимися жидкостями; обычно одной из фаз является вода. Примеры эмульсий: молоко, майонез, маргарин, бензол в воде.
Пены – грубодисперсные системы, состоящие из ячеек, заполненных газом, и отделенных друг от друга пленками очень малой толщины. К пенам относятся: мыльная пена, мусс, пелус, пенопласты.
Характерным признаком грубодисперсных систем является то, что частицы дисперсной фазы видны в обычный микроскоп или даже невооруженным глазом.
Частицы в коллоидных системах уже невозможно различить в обычный микроскоп, так как их размеры меньше длины волны видимого света.
Дисперсные системы с частицами коллоидных размеров называют золями. По характеру дисперсионной среды различают: гидрозоли (растворитель – вода), аэрозоли (мельчайшие капельки жидкости, тонкораспыленные в газе, например, туман), мелкие твердые частицы в газе (дым).
Помимо классификации дисперсных систем по размерам частиц, существует и другая классификация – по агрегатному состоянию дисперсной фазы и дисперсионной среды.
| Дисперсная фаза | Дисперсионная среда | Название системы |
| Жидкость Твердое тело Газ Жидкость Твердое тело Газ Жидкость Твердое тело | Газ Газ Жидкость Жидкость Жидкость Твердое тело Твердое тело Твердое тело | Аэрозоли (туман, дым, пыль) Пены Эмульсии Суспензии Твердые пены Твердые эмульсии Сплавы, тв.золи |
Коллоидные системы
Коллоидные растворы отличаются от истинных рядом свойств:
1. Малой скоростью перемещения ввиду малой скорости диффузии;
2. Повышенной вязкостью;
3. Гетерогенностью.
От грубодисперсных систем они отличаются относительной устойчивостью. Для коллоидов характерна очень развитая поверхность, в связи с чем в коллоидных растворах исключительно важную роль играет адсорбция.
Существует два основных пути получения коллоидных растворов.
1. Дисперсионный – измельчение частиц грубодисперсных систем до размеров, соответствующих коллоидам. При этом используются обычно физические методы с применением различных диспергаторов и коллоидных мельниц.
2. Конденсационный – укрупнение частиц истинных растворов (ионов, молекул) путем их ассоциации до размеров, соответствующих коллоидам. Применяются обычно химические методы: осаждение, гидролиз, окисления-восстановления, нейтрализации.
Например, в результате гидролиза солей железа (III) получают его гидроксид: Fe3+ + 3H2O = Fe(OH)3 + 3H+.
Для повышения устойчивости коллоидов в раствор вводят стабилизаторы (ПАВ).
Строение и заряд коллоидной частицы. Мицелла
Коллоидная частица состоит из ядра, адсорбирующего из окружающей среды ионы одного вида. Эти ионы называют зарядообразующими, их химическая природа близка химической природе ядра коллоидной частицы.
Например,
| золь | Ядро коллоидной частицы | Зарядообразующие ионы |
| AgJ | mAgJ | Ag+ или J- |
| Fe(OH)3 | mFe(OH)3 | Fe3+, Fe(OH)2+ Или Fe(OH)2+ |
Ядро коллоидной частицы с адсорбированными зарядообразующими ионами притягивает к себе из среды ионы противоположного знака (заряда). Зарядообразующие ионы и противоионы гидратированы. Весь этот комплекс и называют коллоидной частицей. В состав коллоидной частицы входит только часть имеющихся в системе противоионов – их называют связанными. Другая часть противоионов остается в дисперсной среде (жидкой фазе). Эти противоионы называют свободными. Они определяют заряд дисперсионной среды. Все сочетание, состоящее из коллоидной частицы и дисперсионной среды (гидратированных свободных противоионов) называют мицеллой. Мицелла – это структурная единица коллоидного раствора.
Примерный состав коллоидных частиц и мицелл золей иодида серебра и гидроксида железа (III)
| золь | Коллоидная частица | мицелла |
| AgJ | [(mAgJ)nAg+(n-x)NO3-yH2O]x+ | {[(mAgJ)nAg+(n-x)NO3-yH2O]x++xNO3-zH2O}0 |
| Fe(OH)3 | [mFe(OH)3nFe3+3(n-x)Cl-yH2O]3x+ | {[mFe(OH)3nFe3+3(n-x)Cl-yH2O]3x+ + 3xCl-zH2O}0 |
Мицелла имеет сложное строение. Пример строения мицеллы Fe(OH)3 приведен ниже.
[mFe(OH)3nFe3+3(n-x)Cl-yH2O]3x+ + 3xCl-
Ядро связанные противоионы свободные противоионы
Адсорбционный слой диффузионный слой
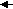
 Коллоидная частица
Коллоидная частица
 Мицелла
Мицелла Суммарно заряд частицы равен разности зарядов адсорбированных ионов и противоионов. Вокруг частицы находится диффузионный слой ионов, заряд которых равен заряду коллоидной частицы. Коллоидная частица и диффузионный слой образуют электронейтральную мицеллу.
Оптические свойства коллоидных растворов
При прохождении через дисперсную систему свет может поглощаться, отражаться или рассеиваться частицами. Поглощение света – явление избирательное. Одни вещества полностью поглощают свет, другие поглощают только лучи определенной части спектра. Отражение света поверхностью частиц возможно только в грубодисперсных системах. Отражение света проявляется в мутности таких дисперсных систем как в проходящем свете, так и при боковом их освещении.
Для типичных коллоидных систем наиболее характерным оптическим свойством является рассеивание света по всем направлениям. Размеры коллоидных частиц меньше длины световой волны, и поэтому рассеивание света обусловлено не отражением его от поверхности частиц, а дифракцией. Рассеивание света при освещении коллоидного раствора было исследовано Тиндалем. Путь света виден при наблюдении сбоку в виде светящейся полосы (конус Тиндаля). Это свечение было названо опалесценцией.
Электрические свойства коллоидных растворов
В 1909 г профессор Московского университета Р. Рейсе наблюдал воздействие постоянного электрического тока на диспергированную в воде глину, и на этом основании описал электрические свойства коллоидных растворов. Частицы дисперсной фазы (глины) перемещались к аноду, где наблюдалось помутнение раствора, а частицы дисперсионной среды (воды) перемещались к катоду, где наблюдалось повышение уровня прозрачной жидкости. Направленное движение частиц к электродам говорило об их заряде. дисперсная фаза несет на себе заряд, противоположный по знаку заряду среды. Движение частиц дисперсной фазы к одному из электродов при пропускании через золь постоянного электрического тока получило название электрофореза, а движение частиц дисперсионной среды – электроосмоса. На границе раздела фаз возникает двойной электрический слой. Между фазами возникает разность потенциалов, называемая электродинамическим потенциалом Ψ. Часть скачка потенциала, обусловленная диффузным слоем, называется электрокинетическим или дзета – потенциалом ξ.
Дзета-потенциал определяется толщиной и зарядом диффузионного слоя, которые зависят от концентрации и заряда противоионов и температуры.
Чем меньше толщина диффузионного слоя, тем больше противоионов этого слоя перейдет за границу скольжения и тем меньше будет значение дзета-потенциала. Чем больше значение ξ, тем больше агрегатная устойчивость коллоидных систем.
Электрические свойства коллоидных растворов объясняют их агрегатную устойчивость, которая проявляется в том, что частицы дисперсной фазы в коллоидном растворе не укрупняются и не слипаются.
Коагуляция коллоидных растворов
Коагуляцией называют процесс соединения коллоидных частиц в крупные агрегаты с последующей потерей коллоидной системой кинетической устойчивости.
Коагуляцию коллоидных растворов можно вызвать нагреванием, охлаждением, интенсивным перемешиванием, а также добавлением различных электролитов. Добавление к коллоидному раствору электролитов приводит к снижению электрокинетического потенциала. Этот процесс характеризуется следующими закономерностями:
- Минимальная концентрация электролита, вызывающая коагуляцию коллоидного раствора, называется порогом коагуляции;
- Коагулирующим действием обладает не весь электролит в целом, а только тот его ион, который имеет заряд, одноименный с зарядом противоионов мицеллы;
- Коагулирующая способность иона зависит от его заряда. Ионы с большим зарядом (например, Fe3+) вызывают коагуляцию при гораздо меньших концентрациях, чем ионы с более низким зарядом (Fe2+);
- Коагулирующая способность ионов с одинаковым зарядом возрастает с увеличением радиуса иона. Ионы органических соединений всегда обладают более высокой коагулирующей способностью, чем неорганических;
- При увеличении концентрации электролита в растворе уменьшается электрокинетический потенциал, и коагуляция наступает при его определенном значении – критическом потенциале.
Коллоидные растворы в природе и технике
В природной воде содержится часть примесей в коллоидном состоянии. Поэтому воду, используемую для коммунальных нужд, электростанций, строительства, подвергают обработке, вызывающей коагуляцию коллоидных частиц.
Коагуляцию широко используют при очистке воды для удаления взвешенных веществ. В качестве коагулянтов используют сульфаты алюминия или железа.
Дымовые газы электростанций, металлургических заводов представляют собой аэрозоли. Для их коагуляции применяется электрогазоочистка методом электрофореза при очень высоких напряжениях. В коллоидном состоянии находятся многие составные части живых организмов: кровь, лимфа, внутриклеточная жидкость.
Наибольшее практическое применение имеют твердые системы с газовой дисперсной фазой, называемые твердыми пенами. На ж/д транспорте применяются такие газонаполненные пластмассы, как минора – для теплоизоляции изотермических, пассажирских и рефрижераторных вагонов, поролон – для изготовления мягких сидений в пассажирских вагонах, стипор - тепло- и звукоизоляционный материал в рефрижераторных вагонах, трубопроводах, стеновых панелях.
Тема: «Окислительно-восстановительные процессы»
План
- Общие понятия и положения. Степень окисления
- Процессы окисления и восстановления
- Составление уравнений, метод электронного баланса
- Типы окислительно-восстановительных реакций (ОВР)
- Направление протекания ОВР
Все ионные реакции в растворах можно разделить на два типа. К первому из них относятся ионообменные реакции, протекающие без изменения заряда ионов, входящих в состав реагирующих веществ. Например, 2NaOH+H2SO4 = Na2SO4 + 2H2O. Ко второму типу относят реакции, идущие с передачей электронов от одних атомов другим – окислительно-восстановительные реакции (ОВР): например, 2KJ+Cl2 = 2KCl+J2.
К окислительно-восстановительным можно отнести реакции с раздельным протеканием окисления и восстановления (электрохимические).
Для характеристики состояния элементов в соединениях введено понятие степени окисления (СО). Под степенью окисления понимают воображаемый (формальный) заряд в соединении, вычисленный, исходя из предположения, что в соединении имеются только ионные связи. СО для большинства соединений имеет условный характер, так как не отражает реальный заряд атома, но, однако, широко используется в химии. В зависимости от электроотрицательности (ЭО) атома СО может быть отрицательной или положительной. Элементы с высоким значением ЭО имеют отрицательную СО, а с малым – положительную.
Определяют СО, исходя из следующих правил:
1. Степень окисления атомов в простом веществе равна нулю. Например, О20, О30, S80 и т.д.
2. В соединениях с ковалентными полярными связями более электроотрицательный элемент имеет следующие СО: F-, О-2 (за исключением пероксидов (Н2О2-), надпероксидов (КО2-1/2), озонидов (КО3-1/3) и O+2F2.
3. Элементы с малой ЭО проявляют СО: Н+ (за исключением солеобразных гидридов LiH-), щелочные металлы +1, щелочноземельные металлы +2.
4. Алгебраическая сумма СО элементов в нейтральной молекуле равна нулю, а в сложном ионе – заряду иона.
Большинство элементов в соединениях проявляют переменную СО.
Примеры. Fe+2O-2, Fe2+3O3-2, K+N+3O2-2, H+N+5O3-2.
Максимальная СО имеет периодическую зависимость от порядкового номера в периодической таблице элементов и совпадает с номером группы. Для неметаллов периодическую зависимость имеет также и минимальная СО – это определяется электронным строением атома.
Окисление – это процесс отдачи электронов атомом. Степень окисления при этом повышается: Zn – 2e = Zn2+.
Вещество, отдающее свои электроны, называется восстановителем (Zn). Zn2+ является окисленной формой цинка.
К типичным восстановителям относят простые вещества, атомы которых имеют низкое значение ЭО, например, металлы (особенно активные – щелочные и щелочноземельные), водород, углерод, анионы, атомы которых находятся в низшей СО, например, Cl-, NO2- и т.д.
Восстановление – это смещение электронов к веществу. СО при этом понижается. Например, Сu2+ + 2e = Cu. Вещество, принимающее электроны (Сu2+), называется окислителем. В данной реакции Cu является восстановленной формой, а Сu2+ - окисленной. К типичным восстановителям относят простые вещества, атомы которых имеют высокую ЭО (кислород, хлор, фтор), соединения кислорода – пероксиды (Н2О2), катионы и анионы, содержащие атомы с высокой СО (Fe3+, Pb4+, NO-3, CrO42-, ClO4-).
В химических реакциях окисление и восстановление – сопряженные процессы. В ходе ОВР восстановитель отдает свои электроны окислителю, сам при этом окисляется. Окислитель, принимая электроны, восстанавливается. Раздельное протекание окисления и восстановления происходит только в электрохимических процессах на электродах.