
Разработка методических основ идентификации избыточных фаз, образующихся в сталях в процессе производства и эксплуатации ответственных изделий машиностроения
| Вид материала | Автореферат |
| надежная идентификация избыточных фаз, образующихся в сталях Дилатометрический метод |
- Разработка методики снижения вреда окружающей среде при обращении с отходами эксплуатации, 359.08kb.
- Реферат по дисциплине " Технологические процессы микроэлектроники " на тему: Технологические, 1398.5kb.
- «Факторы, формирующие качество швейных изделий. Классификация швейных изделий» Йошкар-Ола, 212.16kb.
- «Технология машиностроения» Специализация «Дизелестроение», 37.35kb.
- Разработка рядового керамического кирпича на базе техногенного отощителя с учетом представлений, 7.06kb.
- Руководство по эксплуатации альбом приложений, 259.28kb.
- Планирование выпуска продукции в условиях рынка. Разработка ассортиментной политики, 204.69kb.
- Особенности алгоритмов идентификации образов человеком и компьютером Владимир И. Андреев, 93.61kb.
- Методические рекомендации по применению примерной номенклатуры дел 4 Список использованных, 433.71kb.
- 6М073000 -производства строительных материалов, изделий и конструкции, 145.77kb.
надежная идентификация избыточных фаз, образующихся в сталях;
надежная идентификация и количественное определение содержания фаз, составляющих матрицу.
Количество избыточных фаз, их размер, форма, расположение, химический состав, кристаллическая структура являются во многих случаях определяющими параметрами для свойств сталей. Под идентификацией фаз понимают определение их индивидуальных характеристик (кристаллическая структура, химический состав, размер, форма). К наиболее распространенными методам исследования фазового состава сталей можно отнести следующие:
- оптическая металлография (ОМ);
- рентгеноструктурный анализ;
- просвечивающая электронная микроскопия;
- растровая электронная микроскопия;
- потенциостатический метод.
Кроме перечисленных выше методов существуют иные методы, позволяющие идентифицировать избыточные фазы. К ним можно отнести, например, дифракцию нейтронов, атомно-силовую микроскопию, карбидный анализ, дилатометрию. Однако по совокупности параметров (стоимость, трудоемкость, число получаемых характеристик, сложность интерпретации) перечисленные методы относятся к числу наиболее эффективных.
По результатам представленного аналитического обзора сопоставлены возможности указанных методов идентификации избыточных фаз. Оптическая металлография наиболее эффективна при работе с первичными фазами - неметаллическими включениями. Достаточно большие размеры, хорошо известная морфология позволяют уверенно идентифицировать эти фазы методами оптической металлографии. При работе со вторичными фазами возникают проблемы, обусловленные как размерами, так и необходимостью привлекать первичные признаки – кристаллическую структуру и химический состав. Более информативными при исследовании вторичных фаз, безусловно, являются методы электронной микроскопии, использующие дифракцию электронов и рентгеноспектральный микроанализ. Однако для этих методов негативно сказывается тот факт, что анализ проводится без разделения фаз. Это накладывает ограничения, связанные с присутствием большого количества мелкодисперсных фаз в сталях. Получаемые комплексные картины электронной дифракции от множества частиц очень трудно или даже невозможно расшифровать практически. Полученный при помощи рентгеноспектрального микроанализа элементный состав фаз в таком случае будет некоторым усреднением по нескольким фазам. Рентгеноструктурный фазовый анализ обладает тем преимуществом, что исследует разделенные фазы. Это позволяет, в принципе, получать дифракционные картины с достаточно небольшого числа фаз, что способствует их расшифровке. Таким образом, метод рентгеноструктурного анализа совместно с методами разделения избыточных фаз (по различным параметрам) будет способствовать более надежной идентификации фаз в сталях.
Фазовый состав матрицы сталей также оказывает существенное влияние на свойства металла. В соответствии с этим знание количественного соотношения фаз, составляющих твердый раствор, является важной характеристикой, позволяющей оценить состояние металла в процессе производства и эксплуатации.
К наиболее распространенными методам определения фазового состава матрицы стали можно отнести следующие:
- оптическая металлография;
- магнитные методы (ферритометрия, аустенитометрия);
- дилатометрия;
- рентгеноструктурный фазовый анализ;
- электронная микроскопия (просвечивающая и сканирующая).
Методы можно разделить на методы чисто исследовательские и методы контроля, т.е. методы, доведенные до уровня нормативных документов. К последним относятся рентгеноструктурный фазовый анализ, оптическая металлография и магнитный метод.
Оптическая металлография позволяет разделять присутствующие в матрице фазы по морфологии – размеру, форме, месту расположения, цвету. В настоящее время существует большое число методов анализа изображения при помощи мощных компьютерных программ. Они позволяют с большой степенью достоверности проводить количественный анализ изображений, в том числе и полученных на оптических металлографических микроскопах. На сегодняшний день основной задачей металлографии при анализе фазового состава матрицы сталей является создание условий, при которых частицы разных фаз будут отличаться друг от друга, например, по цвету.
Следует отметить, что процедура, которая используется при выделении различных фазовых компонент твердого раствора, достаточно сложна. Ее конечный результат зависит от большого числа переменных параметров. Надо подчеркнуть, что конкретная методика эффективна для сталей с содержанием легирующих элементов в достаточно узких пределах и термообработанных по определенной схеме. При значительном отклонении состава или режима термообработки методику необходимо существенно изменять.
Проведенный в этой главе анализ показывает, что определение количественных соотношений между фазами матрицы методами ОМ достаточно эффективны, но требуют индивидуального подхода в зависимости от состояния металла. Большое число параметров подготовки металлографического шлифа (способы шлифовки и полировки, компоненты травителя, их соотношения, температура травления, время травления и т.д.) необходимо подобрать для конкретного металла и строго их выдерживать в каждом эксперименте. Кроме того, в ряде случаев применение ОМ затруднено малыми размерами фаз ( 1 мкм).
Достаточно распространенным и доведенным до уровня нормативной документации является магнитный метод определения фаз, составляющих матрицу. Оперативность, относительно невысокая стоимость оборудования, простота процедуры измерения и пробоподготовки, возможность проведения измерения непосредственно на изделии (оборудование, трубопроводы, сварные соединения) делают этот метод незаменимым в производственных условиях в качестве метода контроля. Наличие достаточного числа нормативных документов, касающихся методики измерения, способствует проведению количественного определения фаз матрицы магнитным методом. Однако существует ряд факторов, которые оказывают негативное влияние на результаты, полученные данным методом. К ним относятся морфология частиц ферритной фазы, кристаллическая текстура образца, химический состав, наличие избыточных фаз.
Дилатометрический метод определения количественного соотношения фаз, в том числе и остаточного аустенита, в сталях при различных термообработках используется относительно редко, однако также заслуживает рассмотрения, так как позволяет анализировать фазовый состав непосредственно в процессе термообработки. К недостаткам этой методики, следует отнести невозможность учета влияния образования и распада вторичных фаз, а также изменение коэффициента термического линейного расширения в широком интервале температур.
Из всех приведенных выше методов определения фазового состава матрицы в сталях электронная микроскопия является наиболее информативной. Это относится как к просвечивающей, так и к растровой разновидности метода. И тот и другой обладают очень высокой чувствительностью, которая позволяет выявлять самые незначительные количества искомой фазы.
Электронная микроскопия незаменима в случае очень сложной микроструктуры, состоящей из нескольких компонент с различной морфологией. Она позволяет использовать метод дифракции для выявления кристаллической структуры и метод рентгеноспектрального микроанализа для определения химического состава частицы исследуемой фазы.
Необходимо отметить недостатки метода. Например, в случае использования метода фольг для определения остаточного аустенита (ОА) следует учитывать, что ОА очень чувствителен к уровню напряжений, а распределение напряжений в фольге толщиной порядка 1 мкм существенно отличается от аналогичного распределения в массивном образце. Кроме того, этот метод обладает еще одним очень существенным недостатком – высокой трудоемкостью.
Метод рентгеноструктурного анализа начали использовать для количественного определения фазового состава матрицы сталей лишь немногим позже, чем оптическую металлографию. Основным его преимуществом является тот факт, что это прямой метод. Он имеет дело непосредственно с кристаллической решеткой – гранецентрированной (аустенит) и объемноцентрированной (феррит, мартенсит).
Метод рентгеноструктурного фазового анализа базируется на том, что каждая из составляющих твердый раствор компонент (феррит, мартенсит, аустенит) при облучении рентгеновскими лучами дает свой собственный дифракционный спектр. Количественное значение относительной объемной доли феррита или аустенита может быть определено по этому спектру. Обусловлено это тем, что интегральная интенсивность каждого дифракционного пика той или иной фазы пропорциональна объемной доли этой фазы.
Интегральная интенсивность любого дифракционного пика, получающегося в результате отражения от кристаллографической плоскости с индексами (hkl) пропорциональна объемной доли этой фазы. Для стали, состоящей только из феррита () и аустенита (), соотношение интегральных интенсивностей дифракционных пиков можно выразить как:
Ihkl/ Ihkl = (Rhkl/ Rhkl)( V/V), где:
Ihkl – интегральная интенсивность дифракционного пика (hkl) -фазы;
V – объемная доля -фазы;
К – константа, зависящая от выбора геометрии съемки и излучения;
Rhkl – параметр пропорциональный расчетной интегральной интенсивности.
Его рассчитывают исходя из базовых принципов рентгеноструктурного
анализа.
Приведенное уравнение включает только феррит или мартенсит и аустенит, соответственно, если эти фазы составляют весь объем образца:
V + V = 1
Решая систему из двух уравнений с двумя неизвестными, получаем для объемной доли аустенита:
V = 1/[1+ (I/ I)( R/ R)]
Если определение объемной доли проводится в образце, в котором присутствует значимая объемная доля других фаз, например карбидов, то необходимо учитывать их вклад в общий объем:
V + V + Vc = 1
где Vc – объемная доля карбидов в исследуемом образце.
В этом случае объемная доля аустенита может быть рассчитана по формуле:
V = (1- Vc )/[1+ (I/ I)( R/ R)]
Таким образом, можно учесть вклад карбидов в объем образца. Объемную долю карбидов можно определить с помощью электрохимического выделения или металлографического метода. Однако надо учитывать наложение пиков избыточных фаз на пики аустенита и феррита. Влияние вторичных фаз при наложении будет определяться не только положением дифракционных линий, но и различным количеством этих фаз. Вторым фактором, оказывающим большое влияние на результаты определения фазового состава твердого раствора, является кристаллическая текстура исследуемого образца.
Текстура может возникать в слитках при кристаллизации, в процессе электролитического нанесения металлических покрытий, при обработке металла давлением (ковка, прокатка) и вносить значимую систематическую погрешность в результаты рентгеноструктурного фазового анализа.
Таким образом, можно заключить, что к преимуществам метода рентгеноструктурного фазового анализа должны быть отнесены следующие факторы:
- метод является прямым, опирающимся на определение кристаллической структуры;
- метод достаточно универсален и может быть применен к широкому классу объектов;
- метод является достаточно оперативным, так как не требует длительной подготовки образцов.
Основными проблемами, которые на настоящий момент еще не до конца решены при проведении рентгеноструктурного фазового анализа, являются наличие интенсивной кристаллической текстуры и присутствие избыточных фаз.
Во второй главе изложены сведения о материалах, которые были исследованы в работе, и оборудовании, которое применяли при исследованиях. Поскольку основной целью работы являлась разработка методов исследования фазового состава сталей, то при выборе материала основным требованием было обеспечить присутствие как можно большего числа разнообразных фаз - различной морфологии, химического состава и структуры.
Обоснованием выбора материала служили следующие соображения:
- стали, широко применяющиеся в промышленности или имеющие перспективы для использования;
- стали, принадлежащие к различным структурным классам, в пределах каждого из которых можно изменять фазовый состав;
- возможность проверить каждый предлагаемый метод на нескольких сталях.
Применяя тот или иной тип термообработки и, используя условия длительной эксплуатации, можно получить всю линейку карбидов, характерных для низколегированных сталей – М3С, М7С3, М6С, М23С6, а при соответствующем легировании и спецкарбиды Мо2С, VС. Используя легирование и различные термообработки высокохромистых сталей, можно получить карбиды хрома М23С6 различной дисперсности, а также спецкарбиды ванадия и ниобия. В сталях, содержащих азот, вероятно появление разнообразных нитридов и достаточно простых по структуре (VN, TiN), и с более сложной структурой (VN0,35, Cr2N, CrNbN).
После длительных сроков эксплуатации в высоколегированных сталях, используемых в тепловой энергетике, возможно появление интерметаллидов, например -фазы, фаз Лавеса различного состава. Длительная эксплуатация сталей в коррозионноактивных средах приводит к образованию различного вида соединений железа (окислы, гидроокислы, сульфиды, карбонаты), идентификация которых может способствовать пониманию механизмов коррозионных процессов.
Выбор методов и оборудования для исследования был обусловлен поставленными задачами и объектами исследования. Методы и аппаратура обязаны были соответствовать следующим критериям отбора:
- Обладать необходимыми характеристиками (чувствительностью и разрешающей способностью).
- Обеспечить возможность подтверждения полученных рентгеноструктурным методом результатов как минимум еще одним методом.
Рентгеновские исследования проводили на установке УРС 2.0 (съемка в камерах DSK-60 и РКУ-114) и рентгеновских дифрактометрах. Применяли дифрактометры общего назначения ДРОН-3 и «Rigaku UltraX» (Япония). Исследования методами просвечивающей электронной микроскопии проводили на электронном микроскопе «JEM-100B» (JEOL, Япония) методом одноступенчатых углеродных реплик. Исследования методами растровой электронной микроскопии проводили на растровом электронном микроскопе «JSM-6060» (JEOL, Япония) с энергодисперсионной приставкой «JED – 2300». Исследования проводились как непосредственно со шлифа, так и на углеродных репликах. Кроме того, изучение химического состава образующихся фаз проводили на микроанализаторе «CAMEBAX-MICROBEAM» (Cameca, Франция).
Определение химического состава образцов сталей проводили как методами классической химии, так и спектральными методами. Применялось следующее оборудование:
- анализатор содержания серы и углерода CS (LECO, США);
- атомно-эмиссионный спектрометр «PMI-Master Plus» (WAS, Германия);
- атомно-эмиссионный спектрометр с индукционно-связанной плазмой «OPTIMA 2100» (Perkin Elmer, США);
- атомно-абсорбционный спектрометр 403 (Perkin Elmer, США);
- газоанализатор «ONH-2000» (Eltra, Германия).
Исследования методами оптической металлографии проводили на следующем оборудовании:
- стереомикроскоп CZ 61 с цифровой фотокамерой С5060 (Olympus, Япония);
- компьютерная цифровая система для оптической металлографии на базе микроскопа «NEOPHOT-30» (ЦНИИТМАШ, Россия).
Измерения твердости проводили на твердомерах ТК-2, микротвердомере ПМТ-3 и переносном электронном твердомере «ТЭМП-2». Определение ударной вязкости проводилось на маятниковом копре PSW-300 (MFL, Германия).
Определение содержания -феррита и остаточного аустенита магнитными методами в исследуемых сталях выполняли на ферритометрах объемного типа ФЦ-2 (ЦНИИТМАШ, СССР) и локального типа «Förster», «FERITSCOPE MP30E-S» (Fisher, Германия).
Разделение углеродных фаз при помощи термического окисления осуществлялось на дериватографе OD-103-A (MOM, Венгрия).
В третьей главе изложены основы разработанного метода дифференциального рентгеноструктурного фазового анализа и приведены экспериментальные результаты по применению этого метода к различным сталям.
Рентгеноструктурный фазовый анализ (РСФА) является наиболее распространенным методом анализа фазового состава сталей. Это объясняется наличием необходимого оборудования (рентгеновских установок с фоторегистрацией типа УРС, ИРИС, дифрактометров общего назначения типа ДРОН), относительной оперативностью и большим объемом получаемой информации (идентификация фаз, определение их количества, определение средних размеров их частиц, стехиометрического состава и т.д.).
К факторам, мешающим проведению РСФА, относится достаточно большое количество избыточных фаз, выделяющихся в сталях. Этот фактор влияет на разрешающую способность метода – обилие дифракционных линий может приводить к их множественным наложениям. Кроме того, в большинстве сталей обычно присутствует преобладающая фаза, например, цементит в углеродистых и легированных сталях и M23C6 в высокохромистых сталях. Этот фактор уменьшает чувствительность рассматриваемого метода – слишком мала объемная доля остальных, не преобладающих, фаз.
Автор в течение длительного времени разрабатывал и использовал методы дифференциального РСФА сталей, позволяющие выборочно удалять часть фаз из электролитически выделенных осадков и из коррозионных отложений. Это приводит к повышению чувствительности РСФА благодаря росту объемной доли оставшихся фаз и увеличивает разрешающую способность из-за уменьшения количества дифракционных линий.
Для удаления части фаз в представляемой работе были разработаны и использовались следующие методы:
- химическое растворение отдельных фаз;
- разделение по плотности (весу) с использованием центрифуги;
- магнитное разделение (удаление ферромагнитных фаз);
- разделение по размерам с помощью селективных фильтров;
- применение углеродных реплик;
- окисление отдельных фаз.
В принципе для химического метода разделения реактивы, растворяющие те или иные карбиды и нитриды, хорошо известны. Однако надо учитывать тот факт, что нитриды, карбиды, карбонитриды и т.д., электролитически изолированные из сталей, по химическим свойствам отличаются от аналогичных синтетически полученных чистых соединений. Их химическая стойкость существенно зависит не только от легирования другими элементами, но и от стехиометрического состава, размеров частиц. Поэтому для их разделения химическими методами требуются модифицированные способы.
Для ряда сталей, например, 10ГН2МФА, 10Х9МФБ, Р2МА методы растворения преобладающей фазы использовались с небольшими изменениями (время, температура). В других случаях (стали 10Х12Н3М2Ф, 05Х18Н10АГ10МБ, 08Х18АГ18Ш, 06Х16Н15М2Г2ТФР и др.) были проведены дополнительные эксперименты по разделению фаз химическими методами. Необходимость дополнительных экспериментов была вызвана и тем, что в новых сложнолегированных сталях возникали неизвестные ранее фазы. К их числу относится, например, нитрид ванадия с гексагональной кристаллической решеткой VN0,35 или сложный нитрид хрома и ниобия с тетрагональной кристаллической решеткой CrNbN – при некоторых режимах термообработки они могут быть преобладающими фазами и необходимо их удалять для идентификации других фаз.
Фазовый состав электролитически выделенных осадков, проанализированный различными методами, для всех исследованных сталей приведен в таблице 1. Как видно из таблицы 1, значительное количество фаз, идентифицированных при помощи просвечивающей электронной микроскопии, не выявлялись методом РСФА. Причинами этого могли послужить как недостаточный объем фазы, так и наложение дифракционных пиков от различных фаз. Учитывая физические и химические свойства выделившихся фаз, можно выбрать наиболее эффективные методы их разделения.
В ряде случаев было установлено, что наиболее эффективным является химическое разделение. В результате проведенных исследований были предложены оптимальные варианты. Фазовый состав до и после химического разделения приведен в таблице 2.
Таблица 1. Фазовый состав анодных осадков
| Марка стали | Фазовый состав | |||
| Метод РСФА (анодные осадки) | ПЭМ (метод реплик) | |||
| 10ГН2МФА | Fe3C | Fe3C + Mo2C**+ VC** | ||
| 10Х9МФБ | состав 1 состав 2 состав 3 | М23С6+1 слабая линия М23С6+1 слабая линия М23С6+1 слабая линия | М23С6* + Х** + Х* М23С6* + Х** + Х* М23С6* + Х** + Х* | |
| Р2МА | после длительной эксплуатации восстановительная термообработка | М7С3+М23С6+М6С+ М3С М3С + сл. М23С6 | М7С3+М23С6+М6С+М3С+Мо2С+VС М3С+М23С6*+VC**+Mo2C** | |
| 10Х12Н3М2Ф | режим 1 режим 2 режим 3 | VN0,35 +1 слабая линия М23С6 М23С6 | VN0,35** + М23С6* М23С6* + VN0,35** М23С6*+ VN0,35**+ M3C** | |
| 08Х18АГ18Ш | Cr2N | Cr2N*+VN** | ||
| 10% Cr сталь | М23С6 | М23С6** + М23С6***+ VC** | ||
| ЧС-68 | Ti(C,N) | Ti(C,N)+TiN+Ti2S+CeO+MgO | ||
| ДИ-59 | М23С6 | М23С6+ NbC+-фаза | ||
| 05Х18Н10АГ10МБ Nb – 0,25-0,30% после отпуска Nb – 0,1-0,16% | CrNbN М23С6* | CrNbN**** М23С6*+ CrNbN** | ||
| 08Х18Н12Т | М23С6 + TiN | М23С6 + TiN+X | ||
* - размер зерна 1,0 мкм
** - размер зерна 0,1 мкм
*** - размер зерна 0,01 мкм
**** - выявлены две фракции: 0,1 мкм и 5 мкм
Х – фазу не удалось идентифицировать при помощи микродифракции
Таблица 2. Фазовый состав до и после разделения фаз (химический метод)
| Марка стали | Растворенная фаза | Осадок | ||
| 10ГН2МФА | Fe3C | VC + Mo2C | ||
| 10Х9МФБ | состав 1 состав 2 состав 3 | М23С6 | VC + NbC NbC +VC NbC | |
| Р2МА | после длительной эксплуатации восстановительная термообработка | М7С3+М23С6+М6С+М3С М3С+М23С6 | VC + Mo2C | |
| 10Х12Н3М2Ф режим 1 | VN0,35 | М23С6 | ||
| 10Х12Н3М2Ф режим 2 | М23С6 | VN0,35 | ||
| 10Х12Н3М2Ф режим 3 | VN0,35 | М23С6 | ||
| 10Х12Н3М2Ф режим 3 | М23С6 | VN0,35 | ||
| 08Х18АГ18Ш | Cr2N | VN | ||
| Ломакин | М23С6 | VC | ||
| ДИ-59 | М23С6 | NbC+-фаза | ||
| 05Х18Н10АГ10МБ | М23С6 | CrNbN | ||
| 08Х18Н12Т | М23С6 | TiN+V(C,N) | ||
Как следует из анализа результатов, приведенных в таблице 2, применение рентгеноструктурного анализа в сочетании с химическим разделением фаз позволило идентифицировать все образовавшиеся фазы, в том числе и те, которые не были определены методом ПЭМ.
Для того чтобы подтвердить данные РСФА о разделении фаз, дополнительно проводили определение химического состава осадков до и после процедуры химического разделения. Как выявил анализ химического состава карбидных осадков до и после растворения, при соответствующем подборе растворителей и режима растворения, были удалены только преобладающие фазы. Это можно проиллюстрировать на примере стали 10Х9МФБ (состав 3). После разделения фаз химическим методом (был удален карбид хрома - М23С6) содержание ванадия и ниобия в осадках не уменьшилось, хром исчез, а содержания молибдена упало с 0,15% до 0,03%, т.е. содержание V(C,N) и Nb(C,N) не изменилось, так как они не растворились. Уменьшение количества молибдена объясняется тем, что он входит как в М23С6, так и в V(C,N). Таким образом, в процессе химического разделения происходило удаление только преобладающей фазы, которая препятствовала проведению идентификации всех присутствовавших фаз.
Как следует из сопоставления данных, использование только химического метода разделения в отдельных случаях не позволяло однозначно идентифицировать все выделившиеся фазы. Например, в случае стали 10Х12Н3М2Ф (режим 3) не удалось выявить незначительные количества цементита. Для сталей 10ГН2МФА, Р2МА после химического разделения остаются две фазы, и, в случае преобладания одной из них (Mo2C или VC), это мешает их однозначной идентификации. Близкие химические свойства оставшихся фаз затрудняют их дальнейшее раздельное растворение.
В качестве альтернативы и одновременно дополнения к химическому методу можно использовать различия в физических характеристиках. К таковым относятся размеры частиц, их плотность и магнитные свойства. Различия в размерах можно использовать при разделении с помощью фильтров. Различия в плотности будут приводить к дифференциации фаз по скорости осаждения в жидкости, а ускорить этот процесс можно, используя центрифугирование. Различие в магнитных свойствах скажется при внесении выделенных осадков в магнитное поле.
Плотность исследованных избыточных фаз меняется в широких пределах: от 9,0 г/см3 для Mo2C до 5,4 г/см3 для VC. Размеры фаз также изменяются в значительном интервале: от 0,1 мкм до 10 мкм. Это послужило основанием для использования разделения по плотности и размерам с помощью центрифуги и фильтров. Наличие ферромагнитных (M3C, Fe3O4) и парамагнитных фаз позволяло применить магнитный метод разделения.
Кроме чисто физических методов в работе был применен метод углеродных реплик. Этот метод широко используется в просвечивающей электронной микроскопии. В случае РСФА он оказался полезен своей локальностью (можно проанализировать площадь 1мм2) и избирательностью по размеру (в реплику выделяются преимущественно более мелкие частицы фаз). Метод реплик был успешно использован для исследования металла сварного шва труб из стали SAF 2205 (аналог 02Х22Н5АМЗ) Ø 280,8 мм и выделения дисперсной фракции из стали 10Х13Г12С2Н2Д2Б. Использование экстракционных угольных реплик позволяет проводить рентгеноструктурный фазовый анализ без электрохимического выделения фаз и повышает локальность РСФА.
В таблице 3 приведены результаты применения всех не химических методов разделения.
Таблица 3. Фазовый состав после разделения не химическими методами.
| Марка стали | Метод разделения | Фазовый состав до разделения | Фазовый состав после разделения |
| 10ГН2МФА | химический+ центрифуга центрифуга | Mo2C+ VC Fe3С+Mo2C+ VC | VC** Mo2C** VC** |
| 05Х18Н10АГ10МБ Nb – 0,25-0,30%; Nb – 0,1-0,16% | центрифуга | CrNbN**** М23С6*+ CrNbN** | CrNbN** CrNbN** |
| З5Г2 | магнитное разделение | Fe3O4 | FeСО3+ следы Fe3O4 +следы SiO2 |
| 30ХГМ | магнитное разделение | Fe3O4+ следы SiO2 | FeS+ SiO2+ следы Fe3O4 |
| 10Х12Н3М2Ф | селективные фильтры | М23С6 + VN0,35 | VN0,35**+следы М3С** +следы М23С6** |
| SAF 2205(шов) | углеродные реплики | - | CrN** + Cr2N** |
| 10Х13Г12С2Н2Д2Б | углеродные реплики | NbC*+ NbC** | NbC** |
* - размер зерна 1,0 мкм
** - размер зерна 0,1 мкм
**** - выявлены две фракции: 0,1 мкм и 5 мкм
Как следует из анализа данных таблицы 3, физические методы разделения и метод реплик успешно дополняют, а в ряде случаев и заменяют химический метод и позволяют расширить границы применения дифференциального рентгеноструктурного анализа.
Отдельный раздел работы посвящен проблемам, возникающим при идентификации углеродных фаз, образующихся при синтезе сверхтвердых фаз в металлических матрицах (медь, железо). Необходимость решения указанной задачи возникла в связи с разработкой способа изготовления алмазов методом ударно-волнового нагружения (УВН) порошковой смеси металл-фуллерен.
Основная цель исследований, изложенных в данном разделе – обнаружение и идентификация фаз в компактах разного состава, полученных методом ударно-волнового нагружения, с учетом возможного образования промежуточных метастабильных фаз (гексагональный алмаз и n-алмаз) наряду с кубическим алмазом.

Рис. 1. Углеродные фазы, выделенные из компакта после УВН
F – фуллерен; D – алмаз; G – графит.
Для удаления металлической основы проводили обработку таблеток азотной кислотой. В результате получали только углеродные фазовые составляющие. На рисунке 1 приведено изображение полученных после такой обработки частиц.
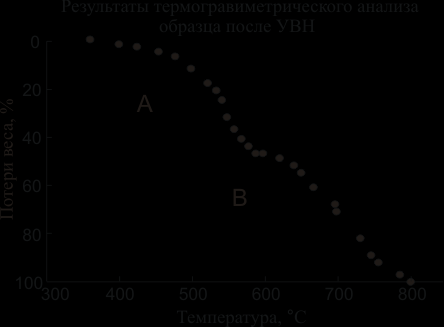
Предварительный анализ показал, что для разделения алмазов и фуллеритов наиболее перспективен метод термического окисления. Были исследованы процесса термического окисления на воздухе продуктов синтеза алмазов, выделенных из компактов, которые были подвергнуты УВН при разных давлениях. С этой целью были проведены термогравиметрические измерения на дериватографе OD-103-A в потоке воздуха при атмосферном давлении в интервале температур 201000С. В качестве образцов сравнения исследовали исходный фуллереновый порошок и порошок ультрадисперсных алмазов.
На рисунке 2 приведена типичная температурная зависимость процентной доли весовых потерь для одного из образцов в состоянии после УВН. Построенная термограмма содержит два участка: низкотемпературный участок А (20590С) и высокотемпературный участок B (600900С). Рентгеноструктурный фазовый анализ образца, нагретого до 590С, не обнаружил в его составе непрореагировавших фуллеритов: дифракционные линии ГЦК С60 полностью исчезли. Это означало, что окисление фуллеренов происходит на участке А, полностью заканчиваясь до 590С. Таким образом, процесс высокотемпературного окисления может быть использован для разделения непрореагировавших фуллеритов и синтезированных алмазов.
А
B
Рис. 2. Термограмма зависимости весовых потерь для образца, нагруженного при 28 ГПа (нагрев в потоке воздуха со скоростью 2,5С)
В главе 4 изложены результаты исследований, посвященных разработке и совершенствованию методов идентификации и определения количественного соотношения фаз, составляющих матрицу стали. Эти методы распространяются как на стали, в которых одна из фаз присутствует в минимальных количествах (остаточный аустенит в мартенситных сталях, -феррит и деформационный мартенсит в аустенитных сталях), так и на стали, в которых фазы присутствуют в одинаковых количествах (дуплексные стали).
Количество остаточного аустенита в сталях является значимым технологическим параметром, влияющим на эксплуатационные свойства продукции машиностроительной отрасли, поэтому его количество является важной, а иногда и определяющей характеристикой при анализе качества продукции. Определение его содержание в сталях на настоящий момент представляет собой серьезную и не до конца решенную задачу. Целью данного раздела работы было изучение возможности его определения с использованием методов рентгеноструктурного анализа.
В предыдущих исследованиях предлагалось либо работать только с нетекстурированными образцами, либо использовать отражение с низкой симметрией для уменьшения влияния текстуры, либо проводить измерения по сумме интенсивностей трех пиков каждой из фаз - и . В первом случае крайне сужается круг объектов, с которыми можно работать. Во втором – влияние текстуры лишь уменьшаться, но не исключается полностью. В третьем – не удастся избежать наложения дифракционных пиков карбидов (избыточных фаз) на все пики - и -фазы. В результате проблема влияния кристаллической текстуры не снимается полностью.
В качестве материала исследования были выбраны стали, позволяющие получить достаточно обширную область содержания ОА – от десятых долей процента до десятков процентов. Материалом исследования послужили образцы:
- стали ШХ15СГ, которые были получены из колец и роликов, изготовленных по штатной технологии (содержание ОА до 20%);
- образцы стали 10Х12Н3М2ФА после закалки с 1050С и отпуска; температуру и продолжительность отпуска варьировали в достаточно широких пределах для изменения фазового состава, в том числе и количества остаточного аустенита от десятых долей процента до нескольких процентов;
- сварного соединения двух секторов труб из стали 06Х12Н3Д, изготовленного для моделирования возможных отклонения от штатной термообработки сварных соединений, выполненных электродами ЦЛ-51.
В стали ШХ15СГ интенсивное формирование кристаллической текстуры происходит в аустенитной области, т.е. до мартенситного превращения. При наличии ориентационного соответствия, характерного для этого превращения, – плоскость (111) аустенита параллельна плоскости (110) мартенсита – кристаллическая текстура не будет влиять на соотношение интегральных интенсивностей дифракционных пиков от указанных плоскостей. Поэтому определение количества остаточного аустенита по пикам (110) и (111) избавлено от влияния кристаллической текстуры.
Измерение интегральных интенсивностей линий (110) и (111) связано с некоторыми затруднениями, поскольку они перекрывают друг друга. Однако использование этих отражений имеет и ряд преимуществ. Во-первых, эти линии являются наиболее интенсивными в дифракционном спектре - и -фазы соответственно. Во-вторых, в ряде случаев именно кристаллографические плоскости (110) и (111) являются составляющими кристаллической текстуры, что позволяет учитывать ее влияние на количественное определение фаз.
По предложенному в ASTM E975 критерию, учитывающему отношение интегральных интенсивностей пиков (211)/(200) и (220)/(200), было установлено, что текстура в этих фазах достаточно интенсивна и пренебречь ею нельзя. Это подтвердили и съемки, проведенные с двух перпендикулярных плоскостей образцов. Они показали, что при использовании линий (110) и (111) количество остаточного аустенита, полученного с использованием этих плоскостей, практически не различалось (не более чем на 0,2%). Использование других дифракционных линий приводило к существенным различиям (до двух раз). Таким образом, использование любых дифракционных линий, кроме (110) и (111) привело бы к недостоверным результатам, обусловленным влиянием кристаллической текстуры.
Для выяснения влияния вторичных фаз (карбидов) на результаты измерения определяли их фазовый состав и содержание в образцах.
Количество определяли весовым методом после электролитического выделения анодных осадков. Фазовый состав исследовали рентгеноструктурным методом на тех же осадках. Единственным карбидом в образцах стали ШХ15СГ оказался цементит – Fe3C, количество которого менялось от 8 % масс. до 10 % масс. в различных образцах.
На рисунке 3 приведена часть дифрактограммы, полученной с кольца, а также отмечены пики цементита, попадающие в эту область. Как видно из рисунка в случае наличия цементита возникают наложения нескольких его дифракционных пиков на пик (110) -фазы и одного пика на пик (111) -фазы. Поскольку все эти линии относятся к числу самых интенсивных в цементите, а количество последнего достаточно велико, в расчетах количества остаточного аустенита необходимо учитывать как наложение пиков, так и присутствие цементита в объеме образцов. Объем, занимаемый цементитом, можно учесть, вводя новый член в соответствующие уравнения:
V = 1/(1+KI/I), где К = Rhkl/ Rhkl (1)
V = (1-Vц)/(1+KI/I ) (2)
где: V - объемная доля -фазы,
V - объемная доля -фазы,
Vц - объемная доля цементита,
Химический состав карбидов оказывает существенное влияние на их рентгеновский дифракционный спектр. От химического состава зависят и положения линий, и их относительная интенсивность. Кроме того, на форму дифракционных пиков влияет и размер частиц карбидов. Исходя из этого, было решено использовать в качестве эталонных спектры карбидов, выделенных электролитически непосредственно из исследуемых образцов. Использование в качестве эталонных иных дифракционных спектров, например, по картотеке JCPDS или PDF-2 могло привести к появлению дополнительной ошибки, связанной с различиями положений и интенсивностей, обусловленными отличиями в химическом составе карбидов и их размерах.
По дифрактограммам, полученным с карбидных осадков образцов, измеряли интегральные интенсивности пиков цементита (103), (022), (210) и (121), лежащие в области пиков (110) и (111). Затем эти интенсивности пересчитывали в соответствии с измеренной интегральной интенсивностью пика (121), полученного при съемке непосредственно с поверхности образца (см. рисунок 3). Интегральные интенсивности линий аустенита и мартенсита корректировались в соответствии с полученными данными: